吡咯并苯并二氮杂二聚物前体及其配体-连接体缀合化合物的制作方法
本发明涉及吡咯并苯并二氮杂二聚物前体及其配体-连接体缀合化合物、包含其的组合物以及尤其作为抗癌剂的治疗应用。
背景技术:
众所周知,吡咯并苯并二氮杂(pyrrolobenzodiazepine,pbd)为通过多种放线菌(actinomycetes)产生的具有抗生或抗肿瘤活性的天然物质。吡咯并苯并二氮杂为序列选择性dna烷基化抗癌剂,与细胞dna共价结合。吡咯并苯并二氮杂为dna交联剂(dna-crosslinkingagents),公知为比全身化疗剂具有显著更强的抗癌活性,在不破坏dna螺旋结构(helix)的情况下,可阻止癌细胞的分裂。
吡咯并苯并二氮杂具有如下所示的一般结构:
上述吡咯并苯并二氮杂在芳香环a及吡咯并c环中的取代基的数量、类型及位置和c环的饱和度方面具有差异。在b环中用于担当dna的烷基化的作为亲电子中心的n10-c11位置存在亚胺(n=c)、甲醇胺(nh-ch(oh))或甲醇胺甲醚(nh-ch(ome))。
一些吡咯并苯并二氮杂二聚物正处于一期临床试验阶段,即,对于急性髓性白血病(aml)疾病以西雅图遗传学公司的sgn-cd123a作为dpbd缀合物来治疗急性髓性白血病(acutemyelocyticleukemia,aml)患者。
已知复星医药(kolltanpharmaceuticals)和泰克/罗氏(genentech/roche)等开发将吡咯并苯并二氮杂作为细胞毒性药物的抗体-药物缀合物。除此之外,spirogen开发基于吡咯并苯并二氮杂的急性髓性白血病治疗剂技术。
与此相关地,有关于吡咯并苯并二氮杂及其缀合物的公开专利(medimune公司、专利文献1)、关于用于治疗增殖性疾病的的非对称吡咯并苯并二氮杂二聚物的公开专利(medimune公司、专利文献2)、关于吡咯并苯并二氮杂的授权专利(medimune公司、专利文献3)、关于用于治疗增殖性疾病的吡咯并苯并二氮杂的授权专利(medimune公司、专利文献4)、关于吡咯并苯并二氮杂的公开专利(medimune公司、专利文献5)、关于吡咯并苯并二氮杂的授权专利(spirogen公司、专利文献6)等。这些公开了改变吡咯并苯并二氮杂化合物结构来提高抗肿瘤活性等,或者仅公开了可以将具有这种变形的结构的吡咯并苯并二氮杂化合物以抗体-药物缀合物的形式给药来提高抗癌活性。
另一方面,具有公开有关对于吡咯并苯并二氮杂二聚物(dimer)的形态具有与氨基甲酸酯相连接的形态的抗体-药物缀合物的技术、将单体形态的吡咯并苯并二氮杂化合物作为前体(prodrug)形态以示出细胞毒性少且稳定的论文、有关n10-(4-硝基苄基)氨基甲酸酯-保护的吡咯并苯并二氮杂前体的制备方法及活性的研究论文(参照非专利文献7、非专利文献8及非专利文献9)。
但是,在上述技术的情况下,合成吡咯并苯并二氮杂时,在如下方面上受限制,即,由于收率低,因此存在不容易扩大的问题,并且存在给药后在血液中的稳定性下降的问题不足以解决。因此,需要开发可提高吡咯并苯并二氮杂的收率的制备方法及用于在给药后提高血液中稳定性并降低毒性的前体化技术。
另一方面,抗体-药物缀合物(adcs,antibody-drugconjugates)为将毒素或药物结合在与抗原相结合的抗体后,在细胞内部释放毒性物质的同时,致使癌细胞凋亡的靶向性新技术。由于以最小限度地影响健康的细胞并将药物准确地传递到靶癌细胞,并且以使仅在特定条件下释放,因此该技术为功效优于抗体治疗剂本身,相对于现有抗癌剂,可大大降低副作用风险的技术。
这种抗体-药物缀合物的基本结构由“抗体-连接体-低分子药物(毒素)”构成。其中,连接体不仅起到简单连接抗体与药物的功能作用,而且,在体内循环中稳定地到达靶细胞后,药物进入细胞内,以使药物通过抗体-药物之间的解离现象(例如,基于酶的水解的结果)容易掉落并对靶癌细胞产生药效。即,根据连接体的稳定性,连接体在抗体-药物缀合物的功效及全身毒性(systemictoxicity)等安全性方面起到重要的作用(discoverymedicine2010,10(53):329-39)。
本发明的发明人开发了一种包含在血浆中更加稳定且在体内循环中也稳定,而且药物在癌细胞中容易释放并可产生药效的有效的自分解基(self-immolativegroup)的连接体,从而对此获得了专利(韩国授权专利第1628872号等)。
现有技术文献
专利文献
(专利文献1)韩国公开专利第2013-0040835号(2013年4月24日公开)
(专利文献2)韩国公开专利第2011-0075542号(2011年6月30日公开)
(专利文献3)韩国授权专利第1700460号(2017年1月20日授权)
(专利文献4)韩国授权专利第1687054号(2016年12月9日授权)
(专利文献5)韩国公开专利第2015-0016245号(2015年2月11日公开)
(专利文献6)韩国授权专利第1059183号(2011年8月18日授权)
(专利文献7)pct/us2016/063564号
(专利文献8)pct/us2016/063595号
(专利文献9)韩国公开专利第2014-0035393号(2014年3月21日公开)
(专利文献10)wo2017/160569(2017年9月21日公开)
(专利文献11)美国专利8697688号(2014年4月15日授权)
(专利文献12)美国专利9713647号(2017年7月25日授权)
(专利文献13)美国公开专利2015-0283258(2015年10月8日公开)
非专利文献
(非专利文献1)kempgarycetal.,synthesisandinvitroevaluationofsg3227,apyrrolobenzodiazepinedimerantibody-drugconjugatepayloadbasedonsibiromycin,bioorganic&medicinalchemistrylettersvol.27no.5,1154-1158(2017)
(非专利文献2)juliamantajetal.,fromanthramycintopyrrolobenzodiazepine(pbd)-containingantibody-drugconjugates(adcs),angewandtechemieinternationaleditionvol.56no.2,462-488(2017)
(非专利文献3)giddensannac.etal.,analoguesofdnaminorgroovecross-linkingagentsincorporatingaminocbi,anaminoderivativeoftheduocarmycins:synthesis,cytotoxicity,andpotentialaspayloadsforantibody-drugconjugates,bioorganic&medicinalchemistryvol.24no.22,6075-6081(2016)
(非专利文献4)hartley,ja,thedevelopmentofpyrrolobenzodiazepinesasantitumouragents,expertopininvdrug,20(6)733-744(2011)
(非专利文献5)kamalahmedetal.,synthesis,anticanceractivityandmitochondrialmediatedapoptosisinducingabilityof2,5-diaryloxadiazole-pyrrolobenzodiazepineconjugates,bioorganic&medicinalchemistryvol.18no.18,6666-6677(2010)
(非专利文献6)guichards.metal.,influenceofp-glycoproteinexpressiononinvitrocytotoxicityandinvivoantitumouractivityofthenovelpyrrolobenzodiazepinedimersjg-136,europeanjournalofcancervol.41no.12,1811-1818(2005)
(非专利文献7)zhang,dongluetal,linkerimmolationdeterminescellkillingactivityofdisulfide-linkedpyrrolobenzodiazepineantibody-drugconjugates,acsmedicinalchemistryletters,7(11),988-993(2016)
(非专利文献8)masterson,lukea.etal.,synthesisandbiologicalevaluationofnovelpyrrolo[2,1-c][1,4]benzodiazepineprodrugsforuseinantibodydirectedenzymeprodrugtherapy,bioorganic&medicinalchemistryletters,16(2),252-256(2006)
(非专利文献9)sangnou,m.j.etal.,designandsynthesisofnovelpyrrolobenzodiazepine(pbd)prodrugsforadeptandgdept,bioorganic&medicinalchemistryletters,10(18),2083-2086(2000)
(非专利文献10)naturerev.cancer2005,5(5),pp.405-12;naturechemicalbiology,2010,17,pp.498-506;lanekt,beesls,structuralbiologyofproteinoffarnesyltransferaseandgeranylgeranyltransferasetypei,journaloflipidresearch,47,pp.681-699(2006);patrickj.kasey,miguelc.seabra;proteinprenyltransferases,thejournalofbiologicalchemistry,vol.271,no.10,issueofmarch8,pp.5289-5292(1996)
(非专利文献11)benjaminp.duckworthetal,chembiochem2007,8,98;uyent.t.nguyenetal,chembiochem2007,8,408;guillermor.labadieetal,j.org.chem.2007,72(24),9291;jamesw.wollacketal,chembiochem2009,10,2934
(非专利文献12)iranm.bell,j.med.chem.2004,47(8),1869
(非专利文献13)berge,etal.,j.pharm.sci.,66,1-19(1977)
技术实现要素:
技术问题
在本发明中,提供给药后血液中稳定性下降的可提高吡咯并苯并二氮杂的血液中稳定性的新型结构的吡咯并苯并二氮杂二聚物前体。
在本发明中,还提供如下的药物前体-连接体-配体系统,即,通过结合包含在血浆中更加稳定且在体内循环中也稳定,药物在癌细胞中易于释放,从而可使药效最大化的自分解基的连接体技术,使上述吡咯并苯并二氮杂二聚物前体稳定地到达靶细胞,可有效发挥药效并大大降低毒性。
解决问题的方案
本发明涉及吡咯并苯并二氮杂二聚物前体(pyrrolobenzodiazepinedimerprodrug)、其药学上可接受的盐或溶剂化物。
更具体地,本发明提供吡咯并苯并二氮杂二聚物前体、其药学上可接受的盐或溶剂化物,在本发明的吡咯并苯并二氮杂二聚物中,
在吡咯并苯并二氮杂二聚物的n10及n'10位置附着有分别独立地选自由-c(o)o*、-s(o)o*、-c(o)*、-c(o)nr*、-s(o)2nr*、-p(o)r'nr*、-s(o)nr*以及-po2nr*基组成的组中的一种,
其中,*为附着有连接体的部分,
其中,r及r'分别独立地为h、oh、n3、cn、no2、sh、nh2、onh2、nhnh2、卤素、取代或未取代的c1-8烷基、取代或未取代的c3-8环烷基、取代或未取代的c1-8烷氧基、取代或未取代的c1-8烷基硫基、取代或未取代的c3-20杂芳基、取代或未取代的c5-20芳基、单-c1-8烷基氨基或二-c1-8烷基氨基,
其中,在c1-8烷基、c3-8环烷基、c1-8烷氧基、c1-8烷基硫基、c3-20杂芳基、c5-20芳基被取代的情况下,被选自由h、oh、n3、cn、no2、sh、nh2、onh2、nnh2、卤素、c1-6烷基、c1-6烷氧基以及c6-12芳基组成的组中的取代基取代。
在本发明的一实施方式中,提供吡咯并苯并二氮杂二聚物前体。在以本发明的前体形式给药的情况下,相对于现有吡咯并苯并二氮杂药物,在以下方面有利,即,由于在血液中暴露时需要通过额外的反应转换为有效物质,因此可预先防止在意外的连接体分解时可能会发生的副作用的可能性,对于正常细胞的毒性降低,药物更加稳定。
并且,当制备抗体-药物缀合物时,在以现有方法制备的抗体-药物缀合物的情况下,存在杂质的含量高、暴露的亚胺基受亲核试剂(nucleophile)攻击而生成不需要的结构的药物的隐患,相反,以本发明的方法制备的抗体-药物缀合物纯度高,因此具有容易分离的优点,相对于现有吡咯并苯并二氮杂或吡咯并苯并二氮杂二聚物,物性进一步提高。
在本发明的一实施方式中,吡咯并苯并二氮杂二聚物前体、其药学上可接受的盐或溶剂化物的特征在于,吡咯并苯并二氮杂二聚物前体具有下述化学式ia或ia'的结构:
[化学式ia]
在上述式中,
虚线表示c1及c2或c2及c3之间存在任意双键,
r1选自由h、oh、=o、=ch2、cn、rm、orm、=ch-rm'=c(rm')2、o-so2-rm、co2rm、corm、卤素以及二卤素(dihalo)组成的组中,
其中,rm'选自rm、co2rm、corm、cho、co2h以及卤素组成的组中,
其中,rm选自取代或未取代的c1-12烷基、取代或未取代的c2-12烯基、取代或未取代的c2-12炔基、取代或未取代的c5-20芳基、取代或未取代的c5-20杂芳基、取代或未取代的c3-6环烷基、取代或未取代的3元至7元杂环基、取代或未取代的3元至7元杂环烷基以及取代或未取代的5元至7元杂芳基组成的组中,
在c1-12烷基、c2-12烯基、c2-12炔基、c5-20芳基、c5-20杂芳基、c3-6环烷基、3元至7元杂环基、3元至7元杂环烷基或5元至7元杂芳基被取代的情况下,
c1-12烷基、c1-12烷氧基、c2-12烯基、c2-12炔基、c5-20芳基、c5-20杂芳基、c3-6环烷基、3元至7元杂环基、3元至7元杂环烷基或5元至7元杂芳基的各氢原子被分别独立地选自由c1-12烷基、c2-12烯基、c2-12炔基、c5-20芳基、c5-20杂芳基、c3-6环烷基、3元至7元杂环基、3元至7元杂环烷基以及5元至7元杂芳基组成的组中的一种以上取代;
r2、r3及r5分别独立地选自由h、rm、oh、orm、sh、srm、nh2、nhrm、nrmrm'、no2、me3sn及卤素组成的组中,
其中,rm及rm'如上所定义;
r4选自h、rm、oh、orm、sh、srm、nh2、nhrm、nrmrm'、no2、me3sn、卤素、取代或未取代的c1-6烷基、取代或未取代的c1-6烷氧基、取代或未取代的c2-6烯基、取代或未取代的c2-6炔基、取代或未取代的c3-6环烷基、取代或未取代的3元至7元杂环烷基、取代或未取代的c5-12芳基、取代或未取代的5元至7元杂芳基、-cn、-no2、-nco、-orn、-oc(o)rn、-oc(o)nrnrn'、-os(o)rn、-os(o)2rn、-srn、-s(o)rn、-s(o)2rn、-s(o)nrnrn'、-s(o)2nrnrn'、-os(o)nrnrn'、-os(o)2nrnrn'、-nrnrn'、-nrnc(o)ro、-nrnc(o)oro、-nrnc(o)nroro'、-nrns(o)ro、-nrns(o)2ro、-nrns(o)nroro'、-nrns(o)2nroro'、-c(o)rn、-c(o)orn以及-c(o)nrnrn'组成的组中,
其中,在c1-6烷基、c1-6烷氧基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c5-12芳基、5元至7元杂芳基被取代的情况下,c1-6烷基、c1-6烷氧基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c5-12芳基、5元至7元杂芳基的各氢原子能够分别独立地被c1-6烷基、c1-6烷氧基、c2-6烯基、c2-6炔基、c3-c6环烷基、3元至7元杂环烷基、c5-10芳基、5元至7元杂芳基、-orp、-oc(o)rp、-oc(o)nrprp'、-os(o)rp、-os(o)2rp、-srp、-s(o)rp、-s(o)2rp、-s(o)nrprp'、-s(o)2nrprp'、-os(o)nrprp'、-os(o)2nrprp'、-nrprp'、-nrpc(o)rq、-nrpc(o)orq、-nrpc(o)nrqrq'、-nrps(o)rq、-nrps(o)2rq、-nrps(o)nrqrq'、-nrps(o)2nrqrq'、-c(o)rp、-c(o)orp或-c(o)nrprp取代,
其中,rn、ro、rp及rq分别独立地选自由h、c1-7烷基、c2-7烯基、c2-7炔基、c3-13环烷基、3元至7元杂环烷基、c6-10芳基及5元至7元杂芳基组成的组中;
在x及x'附着有分别独立地选自由-c(o)o*、-s(o)o*、-c(o)*、-c(o)nr*、-s(o)2nr*、-p(o)r'nr*、-s(o)nr*以及-po2nr*基组成的组中的一种,
其中,*为附着有连接体的部分,
其中,r及r'分别独立地为h、oh、n3、cn、no2、sh、nh2、onh2、nhnh2、卤素、取代或未取代的c1-8烷基、取代或未取代的c3-8环烷基、取代或未取代的c1-8烷氧基、取代或未取代的c1-8烷基硫基、取代或未取代的c3-20杂芳基、取代或未取代的c5-20芳基、单-c1-8烷基氨基或二-c1-8烷基氨基,
其中,在c1-8烷基,c3-8环烷基,c1-8烷氧基,c1-8烷基硫基,c3-20杂芳基,c5-20芳基被取代的情况下,被选自由h、oh、n3、cn、no2、sh、nh2、onh2、nnh2、卤素、c1-6烷基,c1-6烷氧基以及c5-12芳基组成的组中的取代基取代;
y及y'分别独立地选自由o、s及n(h)组成的组中;
r6为取代或未取代的饱和或不饱和c3-12烃链,
其中,其链(chain)可被一个以上的杂原子、nme或取代或未取代的芳香环阻断(interrupt),
其中,其链或芳香环在其链或芳香环的氢原子中的一个以上的位置可由-nh、-nrm、-nhc(o)rm、-nhc(o)ch2-[och2ch2]n-r或-[ch2ch2o]n-r取代,
其中,rm及r分别如上述rm及r所定义,
其中,n为1至12的整数;
r7为h、取代或未取代的c1-6烷基、取代或未取代的c2-6烯基、取代或未取代的c2-6炔基、取代或未取代的c3-6环烷基、取代或未取代的3元至7元杂环烷基、取代或未取代的c6-10芳基、取代或未取代的5元至7元杂芳基、-orr、-oc(o)rr、-oc(o)nrrrr'、-os(o)rr、-os(o)2rr、-srr、-s(o)rr、-s(o)2rr、-s(o)nrrrr'、-s(o)2nrrrr'、-os(o)nrrrr'、-os(o)2nrrrr'、-nrrrr'、-nrrc(o)rs、-nrrc(o)ors、-nrrc(o)nrsrs'、-nrrs(o)rs、-nrrs(o)2rs、-nrrs(o)nrsrs'、-nrrs(o)2nrsrs、-c(o)rr、-c(o)ors或-c(o)nrrrr',
其中,在c1-6烷基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c6-10芳基、5元至7元杂芳基被取代的情况下,c1-6烷基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c6-10芳基、5元至7元杂芳基的各氢原子分别独立地被c1-6烷基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c6-10芳基、5元至7元杂芳基、-ort、-oc(o)rt、-oc(o)nrtrt'、-os(o)rt、-os(o)2rt、-srt、-s(o)rt、-s(o)2rt、-s(o)nrtrt'、-s(o)2nrtrt'、-os(o)nrtrt'、-os(o)2nrtrt'、-nrtrt'、-nrtc(o)ru、-nrtc(o)oru、-nrtc(o)nruru'、-nrts(o)ru、-nrts(o)2ru、-nrts(o)nruru'、-nrts(o)2nruru'、-c(o)rt、-c(o)ort或-c(o)nrtrt'取代,
其中,rr、rr'、rs、rs'、rt、rt'、ru以及ru'分别独立地选自由h、c1-7烷基、c2-7烯基、c2-7炔基、c3-13环烷基、3元至7元杂环烷基、c5-10芳基以及5元至7元杂芳基组成的组中:
[化学式ia']
在上述式中,
r1、r2、r3、r4、r6、r7以及x如上述化学式ia中所定义,
r8选自由h、卤素、取代或未取代的c1-6烷基、取代或未取代的c2-6烯基、取代或未取代的c2-6炔基、取代或未取代的c3-6杂烷基、取代或未取代的3元至7元杂环烷基、取代或未取代的c5-10芳基、取代或未取代的5元至7元杂芳基、-cn、-no2、-nco、-oh、orm、-oc(o)rm、-oc(o)nrmrm'、-os(o)rm、-os(o)2rm、-srm、-s(o)rm、-s(o)2rm、-s(o)nrmrm'、-s(o)2nrmrm'、-os(o)nrmrm'、-os(o)2nrmrm'、-nrmrm'、-nrmc(o)rm、-nrmc(o)orn、-nrmc(o)nrnrn'、-nrms(o)rn、-nrms(o)2rn、-nrms(o)nrnrn'、-nrms(o)2nrnrn'、-c(o)rm、-c(o)orm以及-c(o)nrmrm'组成的组中,
其中,在c1-6烷基、c2-6烯基、c2-6炔基、c3-6杂烷基、3元至7元杂环烷基、c5-10芳基或5元至7元杂芳基被取代的情况下,c1-6烷基、c2-6烯基、c2-6炔基、c3-6杂烷基、3元至7元杂环烷基、c5-10芳基或5元至7元杂芳基的各氢原子分别独立地被c1-6烷基、c2-6烯基、c2-6炔基、c3-6杂烷基、3元至7元杂环烷基、c5-10芳基、5元至7元杂芳基、-orm、-oc(o)rm、-oc(o)nrmrm'、-os(o)rm、-os(o)2rm、-srm、-s(o)rm、-s(o)2rm、-s(o)nrmrm'、-s(o)2nrmrm'、-os(o)nrmrm'、-os(o)2nrmrm'、-nrmrm'、-nrmc(o)rn、-nrmc(o)orn、-nrmc(o)nrnrn'、-nrms(o)rn、-nrms(o)2rn、-nrms(o)nrnrn'、-nrms(o)2nrnrn'、-c(o)rm、-c(o)orm或-c(o)nrmrm'取代,
rm、rm'、rn以及rn'如上述化学式ia中所定义,
za及zb分别独立地为o、n或s,
r12a、r13a以及r14a分别独立地为h、取代或未取代的c1-6烷基、取代或未取代的c2-6烯基、取代或未取代的c2-6炔基、取代或未取代的c3-6环烷基、取代或未取代的3元至7元杂环烷基、取代或未取代的c5-10芳基、取代或未取代的5元至7元杂芳基、-c(o)r15a、-c(o)or15a及-c(o)nr15ar15a',其中,r15a及r15a'如rm所定义,
其中,在c1-6烷基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c5-10芳基、5元至7元杂芳基被取代的情况下,各氢原子被c1-6烷基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环基、3元至7元杂环烷基、c5-10芳基、5元至7元杂芳基、-oro、-oc(o)ro、-oc(o)nroro'、-os(o)ro、-os(o)2ro、-sro、-s(o)ro、-s(o)2ro、-s(o)nroro'、-s(o)2nroro'、-os(o)nroro'、-os(o)2nroro'、-nroro'、-nroc(o)rp、-nroc(o)orp、-nroc(o)nrprp'、-nros(o)rp、-nros(o)2rp、-nros(o)nrprp'、-nros(o)2nrprp'、-c(o)ro、-c(o)oro或-c(o)nroro'取代;
其中,r13a及r14a与附着有它们的原子相结合来能够形成3元至7元杂环基或3元至7元杂环烷基,或者r13a及r14a与附着有它们的原子相结合来能够形成3元至7元杂芳基,
其中,存在于3元至7元杂环基、3元至7元杂环烷基或3元至7元杂芳基的各氢原子分别独立地被c1-6烷基、c2-6烯基、c2-6炔基、c3-6环烷基、3元至7元杂环烷基、c5-10芳基、5元至7元杂芳基、-oro、-oc(o)ro、-oc(o)nroro'、-os(o)ro、-os(o)2ro、-sro、-s(o)ro、-s(o)2ro、-s(o)nroro'、-s(o)2nroro'、-os(o)nroro'、-os(o)2nroro'、-nroro'、-nroc(o)rp、-nroc(o)orp、-nroc(o)nrprp'、-nros(o)rp、-nros(o)2rp、-nros(o)nrprp'、-nros(o)2nrprp'、-c(o)ro、-c(o)oro或-c(o)nroro'取代;
其中、rn、rn'、ro、ro'、rp以及rp'分别独立地选自由h、c1-7烷基、c2-7烯基、c2-7炔基、c3-13环烷基、3元至7元杂环烷基、c5-10芳基以及5元至7元杂芳基组成的组中;
r1'、r2'、r3'、r4'、r5'、r7'以及r8'分别如r1、r2、r3、r4、r5、r7以及r8所定义。
在本发明的一实施方式中,虚线表示c2及c3之间存在双键。
在本发明的一实施方式中,r1选自由取代或未取代的c1-6烷基、取代或未取代的c2-6烯基、取代或未取代的c5-7芳基以及取代或未取代的c3-6杂芳基组成的组中。
在本发明的一实施方式中,r2、r3以及r5分别独立地为h或oh。
在本发明的一实施方式中,r4为c1-6烷氧基,更具体地为甲氧基、乙氧基或丁氧基。
在本发明的一实施方式中,x及x'分别独立地选自由-c(o)o*、-c(o)*以及-c(o)nr*组成的组中,
其中,r分别独立地为h、oh、n3、cn、no2、sh、nh2、onh2、nnh2、卤素、取代或未取代的c1-8烷基或取代或未取代的c1-8烷氧基,其中,在c1-8烷基或c1-8烷氧基被取代的情况下,被h、oh、n3、cn、no2、sh、nh2、onh2、nnh2或卤素取代。
在本发明的一实施方式中,y及y'为o。
在本发明的一实施方式中,r6为取代或未取代的饱和或不饱和c3-8烃链,
其链能够被一个以上的杂原子或取代或未取代的芳香环阻断,
其中,杂原子为o、s或n(h),芳香环为苯、吡啶、咪唑或吡唑,
其中,其链或芳香环在其链或芳香环的氢原子中的一个以上的位置能够被-nhc(o)ch2-[och2ch2]n-r或-[ch2ch2o]n-r取代,
其中,r的定义如上述r所定义,
n为1至6的整数。
在本发明的一实施方式中,提供选自下述的吡咯并苯并二氮杂二聚物前体、其药学上可接受的盐或溶剂化物:
在上述式中,
ro及r'o分别为氧保护基,可能相同或互不相同。
在本发明中,排除如下结构的化合物:
本发明还提供具有下述化学式iia的结构的缀合物、其药学上可接受的盐或溶剂化物:
[化学式iia]
ligand-(l-d)n
在上述式中,
ligand为配体,
l为连接体,
d为如上所述的吡咯并苯并二氮杂二聚物前体,其中,连接体通过如上所述的d的n10位置、n10'位置或n10及n10'位置或者d的x、x'或x及x'与d相结合,
n为1至20的整数。
在本发明的一实施方式中,连接体通过d的n10及n10'位置或者d的x及x'与d相结合。
在本发明的一实施方式中,n为1至10的整数。
本发明还提供具有下述化学式iib或化学式iib'的结构的吡咯并苯并二氮杂二聚物前体-连接体化合物、其药学上可接受的盐或溶剂化物:
[化学式iib]
[化学式iib']
在上述式中,
虚线、r1、r2、r3、r4、r5、r6、r7、x、y、r1'、r2'、r3'、r4'、r5’、r7’、x'、y'、r8、za、zb、r12a、r13a、r14a、r8’、za’、zb’、r12a’、r13a'以及r14a'分别与权利要求2所述的化学式ia及化学式ia'的化合物的定义相同,
xa及xa'为分别独立地为键(bond)或取代或未取代的c1-6亚烷基,其中,在c1-6亚烷基被取代的情况下,被氢、c1-8烷基或c3-8环烷基取代,
g及g'为葡萄糖醛酸(glucuronide)基、半乳糖苷基或其衍生物,
z选自由h、c1-8烷基、卤素、no2、cn、
r8、r9以及r10分别独立地选自由h、c1-8烷基、c2-6烯基以及c1-6烷氧基组成的组中,m为0至12,
n为1至3的整数,在n为2以上的整数的情况下,每个z可能相同或互不相同,
w为-c(o)-、-c(o)nr”-、-c(o)o-、-s(o)2nr”-、-p(o)r”'nr”-、-s(o)nr”-或-po2nr”-,上述r”及r”'分别独立地为h、c1-8烷基、c3-8环烷基、c1-8烷氧基、c1-8烷基硫基、单-c1-8烷基氨基或二-c1-8烷基氨基、c3-20杂芳基或c6-20芳基,
l为选自由支化单元(branchingunit)、连接单元(connectionunit)以及键合单元(bindingunit)组成的组中的一种以上的单元或这些单元的组合,其中,连接单元用于连接w与键合单元、w与支化单元、支化单元与支化单元或支化单元与键合单元,支化单元用于连接连接单元与w或连接单元与另一连接单元,
支化单元为c2-100烯基(其中,烯基的碳原子能够被一个或多个选自由n、o以及s组成的组中的杂原子取代,烯基还能够被一个或多个c1-20烷基取代)、亲水性(hydrophilic)氨基酸、-c(o)-、-c(o)nr””-、-c(o)o-、-(ch2)s-nhc(o)-(ch2)t-、-(ch2)u-c(o)nh-(ch2)v-、-(ch2)s-nhc(o)-(ch2)t-c(o)-、-(ch2)u-c(o)nh-(ch2)v-c(o)-、-s(o)2nr””-、-p(o)r””'nr””-、-s(o)nr””-或-po2nr””-(其中,r””及r””'分别独立地为h、c1-8烷基、c3-8环烷基、c1-8烷氧基、c1-8烷基硫基、单-c1-8烷基氨基或二-c1-8烷基氨基、c3-20杂芳基或c5-20芳基,s、t、u以及v分别独立地为0至10的整数),
连接单元为-(ch2)r(v(ch2)p)q-,其中,r为0至10的整数,p为0至12的整数,q为1至20的整数,v为单键、-o-或s-,
键合单元为
rv为-nh2、n3、取代或未取代的c1-12烷基、c1-12炔基、c1-3烷氧基、取代或未取代的c3-20杂芳基、c3-20杂环基或取代或未取代的c5-20芳基,
其中,在c1-12烷基、c3-20杂芳基、c3-20杂环基或c5-20芳基被取代的情况下,存在于c3-20杂芳基、c3-20杂环基或c5-20芳基的一个以上的氢原子分别独立地被oh、=o、卤素、c1-6烷基、c1-6烷氧基、c2-6烯氧基、羧基、c1-6烷氧羰基、c1-6烷基羰基、甲酰基、c3-8芳基、c5-12芳氧基、c5-12芳基羰基或c3-6杂芳基取代。
在本发明的一实施方式中,xa及xa'分别独立地为键或c1-3烷基。
在本发明的一实施方式中,z选自由h、
r8、r9以及r10分别独立地选自由h、c1-3烷基以及c1-3烷氧基组成的组中,m为1至6。
在本发明的一实施方式中,w为-c(o)-、-c(o)nr”'-或-c(o)o-,其中,r”'为h或c1-8烷基,
l为选自由支化单元(branchingunit)、连接单元(connectionunit)以及键合单元(bindingunit)组成的组中的一种以上的单元或这些单元的组合,连接单元用于连接w与键合单元、w与支化单元、支化单元与支化单元或支化单元与键合单元,支化单元用于连接连接单元与w或连接单元与另一连接单元,
支化单元为c2-8烯基(其中,烯基的碳原子能够被一个或多个选自由n、o以及s组成的组中的杂原子取代,烯基还能够被一个或多个c1-6烷基取代),或亲水性(hydrophilic)氨基酸、-c(o)-、-c(o)nr””-、-c(o)o-、-(ch2)s-nhc(o)-(ch2)t-、-(ch2)u-c(o)nh-(ch2)v-、-(ch2)s-nhc(o)-(ch2)t-c(o)-或-(ch2)u-c(o)nh-(ch2)v-c(o)-(其中,r””为h、c1-8烷基、c3-8环烷基、c1-8烷氧基、c1-8烷基硫基、单-c1-8烷基氨基或二-c1-8烷基氨基、c3-20杂芳基或c5-20芳基,s、t、u以及v分别独立地为0至5的整数),
连接单元为-(ch2)r(v(ch2)p)q-,其中,r为0至10的整数,p为0至12的整数,q为1至20的整数,v为单键或-o-,
键合单元为
其中,连接单元为-(ch2)r(v(ch2)p)q-,
其中,r为0至8的整数,p为1至12的整数,q为1至10的整数,v为单键或-o-。
在本发明的一实施方式中,g及g'分别独立地可以为β-葡萄糖醛酸基、半乳糖苷基或其衍生物。
在本发明的一实施方式中,提供下述化学式iic的吡咯并苯并二氮杂二聚物前体-连接体化合物、其药学上可接受的盐或溶剂化物:
[化学式iic]
在上述式中,
虚线表示c1及c2或c2及c3之间存在任意双键,
r1选自由甲基、乙基、亚甲基、甲氧基以及取代或未取代的苯基组成的组中,在苯基被取代的情况下,被选自由h、oh、卤素、c1-6烷基、c1-6烷氧基以及c6-12芳基组成的组中的取代基取代,
m为1至10的整数,
n为1至10的整数。
在本发明的一实施方式中,在上述化学式iic中,r1为甲基、亚甲基;以及可以为被选自由h、oh、卤素、c1-6烷基以及c1-6烷氧基组成的组中的取代基取代或未取代的苯基。
在本发明的一实施方式中,在上述化学式iic中,m为2至8的整数,具体为3至7的整数,更具体地可以为4至6的整数。
在本发明的一实施方式中,在上述化学式iic中,n为2至8的整数,具体为3至7的整数,更具体地可以为4至6的整数。
本发明还提供具有下述化学结构的吡咯并苯并二氮杂二聚物前体-连接体化合物、其药学上可接受的盐或溶剂化物。但是,下述吡咯并苯并二氮杂二聚物前体-连接体化合物为例示,本技术领域的普通技术人员可在如上所述的技术范围内可制备及使用多种吡咯并苯并二氮杂二聚物-连接体化合物:
本发明还提供具有下述化学式iiia或化学式iiib的结构的吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物、其药学上可接受的盐或溶剂化物:
[化学式iiia]
[化学式iiib]
在上述式中,
虚线、r1、r2、r3、r4、r5、r6、r7、x、y、r1'、r2'、r3'、r4'、r5'、r7'、x'、y'、r8、za、zb、r12a、r13a、r14a、r8'、za'、zb'、r12a'、r13a'以及r14a'与权利要求2所述的化学式ia及化学式ia'的化合物的定义相同,
xa、g、z、w、l、xa'、g'、z'分别与权利要求11所述的化学式iib的化合物定义相同;
ligand为抗原结合部位。
在本发明的一实施方式中,ligand为蛋白质。
在本发明的一实施方式中,上述蛋白质为寡肽、多肽、抗体、抗原性多肽片段或人工抗体(repebody)。
在本发明的一实施方式中,上述蛋白质具有可以被类异戊二烯转移酶识别的一个以上的氨基酸基序。即,蛋白质的c-末端(片段、其类似物或衍生物)能够结合在可被类异戊二烯转移酶识别的氨基酸基序。
在本发明的一实施方式中,在上述蛋白质与氨基酸基序之间还可包含由氨基酸、寡肽或多肽构成的间隔单元。
在本发明的一实施方式中,上述蛋白质通过氨基酸基序与连接体共价结合。
在本发明的一实施方式中,上述氨基酸基序可与蛋白质的c-末端共价结合或者与至少一种共价结合在蛋白质的c-末端的间隔单元共价结合。蛋白质可直接与氨基酸基序共价结合或者通过与间隔单元共价结合来与氨基酸基序相连接。上述氨基酸间隔单元由1个至20个氨基酸构成,其中,优选为甘氨酸(glycin)单元。
在本发明的一实施方式中,上述蛋白质的c-末端为抗体的轻链或重链。
在本发明的一实施方式中,上述蛋白质为单克隆抗体。
在本发明的一实施方式中,上述类异戊二烯转移酶包括法尼基蛋白转移酶(farnesylproteintransferase,ftase)或香叶烯基转移酶(geranylgeranyltransferase,ggtase),它们分别伴随向法呢基或叶草基-叶草基残基的靶蛋白质的(多个)c-末端半胱氨酸的转移。香叶烯基转移酶可分为香叶烯基转移酶i及香叶烯基转移酶ii。法尼基蛋白转移酶及香叶烯基转移酶i可识别caax基序。
在本发明的一实施方式中,上述氨基酸基序为cyyx、xxcc、xcxc或cxx,其中,c为半胱氨酸,y为脂肪族氨基酸,x为决定类异戊二烯转移酶的底物特异性的氨基酸。
在本发明一实施例中,具有上述氨基酸基序的蛋白质选自由a-hc-(g)zcvim、a-hc-(g)zcvll、a-lc-(g)zcvim以及a-lc-(g)zcvll组成的组中,上述a表示抗体,hc表示重链,lc表示轻链,g表示甘氨酸单元,z为0至20的整数。
类异戊二烯转移酶不仅可以识别异底物(isosubstrate),而且还可以识别底物。异底物是指对底物被变形的底物类似物。类异戊二烯转移酶在蛋白质的c-末端中使特定氨基酸基序(例如:caax基序)烷基化(参照:benjaminp.duckworthetal,chembiochem2007,8,98;uyent.t.nguyenetal,chembiochem2007,8,408;guillermor.labadieetal,j.org.chem.2007,72(24),9291;jamesw.wollacketal,chembiochem2009,10,2934)。官能化蛋白质在(多个)c-末端半胱氨酸中可通过烷基化使用类异戊二烯转移酶及异底物来生成。
例如,c-末端caax基序的半胱氨酸残基可通过使用类异戊二烯转移酶来与异底物进行反应。在特定情况下,aax接着可被蛋白酶去除。所获得的半胱氨酸接着可由酶在羧基末端被甲基化(参照:iranm.bell,j.med.chem.2004,47(8),1869)。
本发明的蛋白质可使用本技术领域中公知的任何分子生物或细胞生物法来制备。例如,可使用瞬时转染法。对可被类异戊二烯转移酶识别的特定氨基酸基序进行编码的基因序列可通过使用标准聚合酶链式反应(pcr)技术被插入至公知的质粒载体,以使在其c-末端表达具有特定氨基酸基序的蛋白质(片段或其类似物)。像这样,可表达具有可被类异戊二烯转移酶识别的一个以上的氨基酸基序的蛋白质。
在本发明的一实施方式中,在蛋白质为单克隆抗体的情况下,单克隆抗体的一个以上的轻链、单克隆抗体的一个以上的重链或两者可包含具有可被类异戊二烯转移酶识别的氨基酸基序的氨基酸位点,本技术领域的技术人员可立即选择使感兴趣的靶选择性结合的蛋白质(例如:受试者的靶细胞)。
在本发明的一实施方式中,可包含与感兴趣的靶特异性结合的抗体或抗原片段。
在本发明的一实施方式中,上述氨基酸基序为cyyx、xxcc、xcxc或cxx(其中,c为半胱氨酸,y为脂肪族氨基酸,x为决定类异戊二烯转移酶的底物特异性的氨基酸),优选地,上述氨基酸基序为cyyx。
本发明还提供包含上述的吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物、其药学上可接受的盐或溶剂化物的用于预防或治疗增殖性疾病的药学组合物。
本发明还提供用于预防或治疗增殖性疾病的药学组合物,包含:吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物、其药学上可接受的盐或溶剂化物;以及药学上可接受的赋形剂的。
本发明还提供用于预防或治疗增殖性疾病的药学组合物,包含:吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物、其药学上可接受的盐或溶剂化物;一种以上的治疗共作用剂(therapeuticco-agent);以及药学上可接受的赋形剂。
在本发明的一实施方式中,上述治疗共作用剂可以为对增殖性疾病具有预防、改善或治疗效果的作用剂、可减少给药增殖性疾病治疗剂时出现的副作用的表达的作用剂或具有免疫力提高效果的作用剂等,但并不限定于此,是指只要是当与吡咯并苯并二氮杂一起以配合剂的形态适用时,具有治疗上有用的效果、和/或进一步提高吡咯并苯并二氮杂的稳定性、和/或降低给药吡咯并苯并二氮杂时出现的副作用、和/或通过提高免疫力来使治疗效果最大化的制剂,可配合任何一种来适用。
在本发明的一实施方式中,上述增殖性疾病是指无论是在试管内或生物体内,不合需要的过度或异常的细胞不会受到不希望的控制的细胞增殖相关疾病,如新生物或过形成性生长。增殖性疾病可选自由新生物、肿瘤、癌、白血病、干癣、骨病、纤维化增生性疾病以及动脉粥样硬化组成的组中。作为新生物及肿瘤的例,可列举组织细胞瘤、神经胶质瘤、星形细胞瘤、骨瘤等。
在本发明的一实施方式中,癌可选自由肺癌、小细胞肺癌、胃肠癌、大肠癌、肠癌、乳腺癌、卵巢癌、前列腺癌、睾丸癌、肝癌、肾癌、膀胱癌、胰腺癌、脑癌、肉瘤、骨肉瘤、卡波西肉瘤以及黑色素瘤组成的组中,但是,吡咯并苯并二氮杂只要能够产生治疗效果的癌瘤,均可适用。
本发明还提供具有增殖性疾病的对象体中的增殖性疾病的治疗方法,包括向对象体给药有效量的用于治疗增殖性疾病的吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物、其药学上可接受的盐或溶剂化物的步骤。
在本发明的一实施方式中,提供包括向患者给药上述药学组合物的步骤的癌治疗方法。
本发明适合使用于向对象体的靶位置提供吡咯并苯并二氮杂化合物。本发明的缀合物释放完全没有包含连接体部分的活性吡咯并苯并二氮杂化合物,不影响吡咯并苯并二氮杂化合物的反应性。
定义
在本说明书中适用如下定义:
在本说明书中,“缀合物(conjugates)”是指与细胞毒性化合物的一个以上的分子共价结合的细胞结合剂。其中,“细胞结合剂”为对生物靶标具有亲和度的分子,例如,为配体、蛋白质、抗体,具体可以为单克隆抗体、蛋白质或抗体片段、肽、寡核苷酸、低聚糖,结合剂具有将生物学活性化合物诱导至生物靶标的功能。在本发明的一实施方式中,缀合物能够以通过细胞表面抗原来靶向化肿瘤细胞的方式设计而成。抗原可以为从异常细胞类型中过表达或表达的细胞表面抗原。具体地,靶抗原可以为仅在增殖性细胞(例如肿瘤细胞)上表达的。靶抗原通常可以基于增殖性组织及正常组织之间的不同表达来选择。在本发明中,配体与连接体相连接。
在本说明书中,“抗体”是通过位于免疫球蛋白分子的可变区域的至少一个抗原识别部位靶标,例如,与碳水化物、多核苷酸、脂质、多肽等能够特异性结合的免疫球蛋白分子。在本说明书中所使用的术语“抗体”不仅包括无损伤多克隆或单克隆抗体,而且还包括拥有与规定的抗原特异性结合的能力的无损伤抗体的任意抗原结合部位(例如,“抗原-结合片段”)或其单链、包含抗体的融合蛋白质及包含抗原识别部位的免疫球蛋白分子的任意其他变形的构型,例如,非限制性地,fab;fab';f(ab')2fd片段;fv片段;单域抗体(dab)片段;单离的互补性决定区(cdr);单链(scfv)以及单域抗体(例如,鲨鱼及骆驼类抗体)、最大抗体、微抗体、内抗体、双抗体、三抗体、四抗体、v-nar以及双-scfv(例如,参考文献[hollingerandhudson,2005,naturebiotechnology23(9):1126-1136])。
抗体包括任意类型的抗体,例如,免疫球蛋白g(igg)、免疫球蛋白a(iga)或免疫球蛋白m(igm)(或其子类型),抗体不必是任意特定类型。根据抗体的重链的恒定区的氨基酸序列,可将免疫球蛋白分配到不同类型。存在五种主要类型的免疫球蛋白:免疫球蛋白a、免疫球蛋白d(igd)、免疫球蛋白e(ige)、免疫球蛋白g(igg)以及免疫球蛋白m(igm),其中,几种可进一步被分为子类型(亚型),例如,免疫球蛋白g1、免疫球蛋白g2、免疫球蛋白g3、免疫球蛋白g4、免疫球蛋白a1以及免疫球蛋白a2。相应于不同类型的免疫球蛋白的重链(hc)恒定域分别被称为α、δ,ε、γ以及μ。不同类型的免疫球蛋白的亚单元结构及三维配位是众所周知的。本发明的抗体可通过利用相关技术领域中公知的技术,例如,重组技术、噬菌体呈现技术、合成技术或上述技术的组合或相关技术领域中容易知道的其他技术来制备。
在本说明书中“单离的抗体”是指实际上并不包含具有不同的抗原特异性的其他抗体的抗体,实际上可能不包含其他细胞物质和/或化学物质。
在本说明书中“生物靶标”是指位于肿瘤、癌细胞、细胞间质(extracellularmatrix)表面的抗原。
在本说明书中“连接体”是指使细胞毒性化合物与配体共价结合的化合物。在本发明的一实施方式中,连接体可使用在pct/us2016/063564号及pct/us2016/063595号中公开的连接体。
在本说明书中“治疗剂”为对增殖性疾病,例如,癌细胞或活化的免疫细胞起到细胞毒性、细胞增殖抑制和/或免疫调节效果的作用剂。作为治疗剂的例,包括细胞毒性剂、化学治疗剂、细胞增殖抑制剂以及免疫调节剂。
在本说明书中“化学治疗剂”为对癌的治疗有用的化学化合物。
在本说明说中,“对象体”是指人类或非人类动物,尤其包括哺乳动物。作为对象体的例,可列举人类对象体,例如,是包括在本说明书中所记载的具有障碍,更具体地为具有癌的人类患者或者正常对象体的概念。“非-人类动物”包括脊椎动物,例如,非-哺乳动物(例如,鸡、两栖类、爬虫类)及哺乳动物,例如,非-人类灵长类、家畜和/或对农业有用的动物(例如,羊、狗、猫、牛、猪等)以及啮齿类(例如,小鼠、大鼠、仓鼠、豚鼠等)。在特定实例中,对象体为人类患者。
在本说明书中“治疗”或“进行治疗”均指治疗性治疗及预防学或预防措施。需要治疗的对象体包括已患疾病的对象体及易患疾病的对象体或需预防疾病的对象体。因此,使用于疾病或需要治疗的对象体的情况下,相对于未处理对象体,上述术语包括阻止或减缓疾病的进展、预防症状、降低疾病和/或症状的严重程度或缩短疾病持续时间,但并不限定于此。
在本说明书中“给药”或“进行给药”是指通过任意适当的途径来提供和/或接触和/或传递化合物或多种化合物,以实现所期望的效果。给药途径可包括通过口服、舌下、非口服(例如,静脉内、皮下、皮内、肌肉内、关节内、动脉内、滑膜内、胸骨内、脊髓内、病变内或颅内注射)、经皮、局部、颊侧、直肠、膣、鼻腔、眼科、吸入以及移植物的给药,但并不限定于此。
在本说明书中“未取代或取代的”是指将可未取代或可取代的的母体基团(parentgroup),“取代的”是指具有一个以上的取代基的母体基团,“取代基”是指与母体基团共价结合或融合于母体基团的化学部分。
在本说明说中“卤素”是指氟、氯、溴、碘等。
在本说明书中“烷基”为从脂肪族或脂环族、饱和或不饱和(不饱和、完全不饱和)烃化合物的碳原子去除氢原子来获得的一价部位,作为饱和烷基的例有甲基、乙基、丙基、丁基、戊基、己基、庚基等,作为饱和直链型烷基的例有甲基、乙基、正丙基、正丁基、正戊基(戊基)、正己基、正庚基等,作为饱和支链型烷基的例,可列举异丙基、异丁基、仲丁基、叔丁基、异戊基、新戊基等。
在本说明书中,“烷氧基”是指-or[其中,r为烷基],可列举甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、异丁氧基、叔丁氧基等。
在本说明书中,“芳基”是指从具有环原子的芳香族化合物的芳香环原子去除氢原子来获得的一价部位。
在本说明书中,“烯基”为具有一个以上的碳-碳双键的烷基,作为不饱和烯基的例,可列举乙烯基(乙烯基,-ch=ch2)、1-丙烯(-ch=ch-ch3)、2-丙烯、异丙烯、丁烯基、戊烯基、己烯基等。
在本说明书中,“炔基”为具有一个以上的碳-碳三键的烷基,作为不饱和炔基的例可列举乙炔基及2-丙炔基等。
在本说明书中,“羧基”是指-c(=o)oh。
在本说明书中,“甲酰基”是指-c(=o)h。
在本说明书中,“芳基”涉及从芳香族化合物的芳香环原子去除氢原子来获得的的一价部位。例如,“c5-7芳基”为部位具有5个至7个环原子,是从芳香族化合物的芳香环原子去除氢原子来获得的一价部位,“c5-10芳基”为部位具有5个至10个环原子,是从芳香族化合物的芳香环原子去除氢原子来获得的一价部位。其中,前缀(c5-7,c5-10等)是指环原子的数或环原子数的范围,而与是否为碳原子还是杂原子无关。例如,“c5-6芳基”涉及具有5个或6个环原子的芳基。其中,环原子可均为碳原子,如“羧芳基”。作为羧芳基的例,包括衍生自苯、萘、薁、蒽、菲、萘并萘以及芘的,但并不限定于此。作为包含至少一个为芳香环的融合环的芳基的例,包括衍生自茚满、茚、异茚、四氢萘、苊、芴、萉、醋菲以及醋蒽的基团,但并不限定于此。或者,环原子可包含一个以上的杂原子,如“杂芳基”。
在本说明书中,“杂芳基”为包含一个以上的杂原子的芳基,例如,吡啶、嘧啶、苯并噻吩、呋喃基、二氧杂丙基、吡咯、恶唑、吡啶基、哒嗪、嘧啶基等,更具体地,可列举衍生自苯并呋喃、异苯并呋喃、吲哚、异吲哚、吲嗪、吲哚啉、异吲哚啉、嘌呤(腺嘌呤或鸟嘌呤)、苯并咪唑、吲唑、苯并恶唑、苯并异恶唑、苯并二茂、苯并呋喃、苯并三唑、苯并噻呋喃、苯并噻唑、苯并噻二唑的具有两个融合环的c9、衍生自吡喃、异吡喃、色满、异色满、苯并二恶烷、喹啉、异喹啉、喹嗪、苯并恶嗪、苯二氮、吡啶并吡啶、喹喔啉、喹唑啉、噌啉、酞嗪、萘啶、蝶啶的具有两个融合环的c10、衍生自苯二氮卓的具有两个融合环的c11、衍生自咔唑、二苯并呋喃、二苯并噻吩、咔啉、哌啶、吡啶并吲哚的具有三个融合环的c13、衍生自吖啶、氧杂蒽、硫杂蒽、氧蒽、吩恶噻、吩嗪、吩恶嗪、吩噻嗪、噻蒽、菲啶、邻菲咯啉、吩嗪的具有三个融合环的c14。
在本说明书中,“环烷基”是作为环基的烷基,涉及从环(cyclic)烃化合物的脂环族环原子去除氢原子来获得的一价部位。作为环烷基的例,包括衍生自以下的,但并不限定于此:
饱和单环烃化合物:环丙烷、环丁烷、环戊烷、环己烷、环庚烷、甲基环丙烷、二甲基环丙烷、甲基环丁烷、二甲基环丁烷、甲基环戊烷、二甲基环戊烷以及甲基环己烷;
不饱和单环烃化合物:环丙烯、环丁烯、环戊烯、环己烯、甲基环丙烯、二甲基环丙烯、甲基环丁烯、二甲基环丁烯、甲基环戊烯、二甲基环戊烯以及甲基环己烯;以及
饱和杂环烃化合物:降蒈烷、降蒎烷、降莰烷。
在本说明书中,“杂环基”涉及从杂环化合物的环原子去除氢原子来获得的一价部位。
在本说明书中,前缀(例如,c1-12、c3-8等)是指环原子的数或环原子的数的范围,而与是否为碳原子还是杂原子无关。例如,在本说明书中,所使用的术语“c3-6杂环基”涉及具有3个至6个环原子的杂环基。
作为单环杂环基的例,包括衍生自以下的,但并不限定于此:
n1:氮丙啶、吖丁啶、吡咯啶、吡咯啉、2h-或3h-吡咯、哌啶、二氢吡啶、四氢吡啶、吖庚因;
n2:咪唑啉啶、吡唑啶、咪唑啉、吡唑啉、哌嗪;
o1:环氧乙烷、氧杂环丁烷、氧戊环、呋喃、恶烷、二氢吡喃、吡喃、恶庚因;
o2:二氧戊环、二恶烷以及二氧杂环庚烷;
o3:三恶烷;
n1o1:四氢唑、二氢唑、四氢异唑、二氢异唑、吗啉、四氢恶嗪、二氢恶嗪、恶嗪;
s1:硫杂环丙烷、硫化环丙烷、硫杂环戊烷、硫化环戊烷、硫杂环庚烷;
n1s1:噻唑啉、噻唑烷、硫吗啉;
n2o1:噁二嗪;
o1s1:氧硫杂环戊烯、氧硫杂环己烷;以及
n1o1s1:恶噻嗪。
在本说明书中,“前体”是指在体内生理条件下(例如,酶氧化(enzymaticoxidation)、还原(reduction)和/或水解等)通过酶、胃酸的作用可直接或间接转换为吡咯并苯并二氮杂药物的化合物。
在本说明书中,作为“药学上可接受的盐”,可使用通过游离酸(freeacid)形成的酸加成盐,作为上述游离酸,可使用有机酸或无机酸。
上述有机酸并不限定于此,包括柠檬酸、醋酸、乳酸、酒石酸、马来酸、富马酸、甲酸、丙酸、草酸、三氟乙酸、苯甲酸、葡萄糖酸、甲磺酸、乙醇酸、琥珀酸、4-甲苯磺酸、谷氨酸以及天冬氨酸。并且,上述无机酸并不限定于此,包括盐酸、溴酸、硫酸以及磷酸。
例如,在化合物具有阴离子或可能为阴离子的官能团的情况下(例如-cooh可以为-coo-),可利用适当的阳离子来形成盐。作为适当的无机阳离子的例,包括:碱金属离子,如na+及k+;碱土金属阳离子,如ca2+及mg2+;以及其他阳离子,如al3+,但并不限定于此。作为适当的有机阳离子的例,包括铵离子(即,nh4+)及取代的铵离子(如nh3r+、nh2r2+、nhr3+、nr4+),但并不限定于此。
作为一些适当的取代的铵离子的例有衍生自以下的:乙胺、二乙胺、二环己胺、三乙胺、丁胺、乙二胺、乙醇胺、二乙醇胺、哌嗪、苄胺、苯基苄胺、胆碱、葡甲胺以及三乙醇胺,还有氨基酸,如赖氨酸及精氨酸。作为通常的季铵离子的例有n(ch3)4+。
在化合物具有阳离子或可能为阳离子的官能团的情况下(如-nh2可以为-nh3+),可利用适当的阴离子来形成盐。作为适当的无机阴离子的例,包括衍生自下述无机酸的,但并不限定于此:可列举盐酸、氢溴酸、氢碘酸、硫酸、亚硫酸、硝酸、亚硝酸、磷酸以及亚磷酸等。
作为适当的有机阴离子的例,包括衍生自下述有机酸的,但并不限定于此:可列举2-乙酰氧基苯甲酸、乙酸、抗坏血酸、天冬氨酸、苯甲酸、樟脑磺酸、肉桂酸、柠檬酸、依地酸、乙二磺酸、乙磺酸、富马酸、葡庚糖酸、葡萄糖酸、谷氨酸、乙醇酸、羟基马来酸、羟基萘羧酸、羟乙磺酸、乳酸、乳糖酸、月桂酸、马来酸、苹果酸、甲磺酸、粘液酸、油酸、草酸、棕榈酸、双羟萘酸、泛酸、苯基乙酸、苯磺酸、丙酸、丙酮酸、水杨酸、硬脂酸、琥珀酸、磺胺酸、酒石酸、甲苯磺酸以及戊酸等。作为适当的聚合物有机阴离子的例,包括衍生自下述聚合物酸的,但并不限定于此:可列举单宁酸、羧甲基纤维素等。
在本说明书中,“溶剂化物(solvate)”是指本发明的化合物与溶剂分子(solventmolecules)之间的分子复合物(molecularcomplex),作为溶剂化物的例,包括与水、异丙醇、乙醇、甲醇、二甲基亚砜(dimethylsulfoxide)、乙酸乙酯、乙酸、乙醇胺或其混合溶剂相结合的本发明的化合物,但并不限定于此。
制备、纯化和/或处理相当于活性化合物的溶剂化物可能方便或理想。术语“溶剂化物”在本说明书中以常规的含义使用,是指溶质(如,活性化合物、活性化合物的盐)及溶剂的络合物。在溶剂为水的情况下,可将溶剂化物方便地成为水合物,如一水化物、二水化物、三水化物等。
上述本发明的药学组合物可包含药学上可接受的载体。通常,药学上可接受的载体可包括缓慢代谢的巨大分子,例如,蛋白质、多糖类、聚乳酸、聚乙醇酸、聚合氨基酸、氨基酸共聚物、脂质凝聚物等,本技术领域的技术人员可适当选择使用这些药学上可接受的载体。
包含药学上可接受的载体的上述组合物可以为口服或非口服的各种剂型。在进行制剂化的情况下,通过使用通常使用的填充剂、增量剂、结合剂、湿润剂、崩解剂、表面活性剂等稀释剂或赋形剂来制备。
用于口服给药的固体制剂包括片剂、丸剂、散剂、颗粒剂、胶囊剂等,这些固体制剂通过混合至少一种以上的赋形剂,如淀粉、碳酸钙、蔗糖或乳糖、明胶等来制备。并且,除了简单的赋形剂之外,还可使用润滑剂,如硬脂酸镁、滑石等。
作为用于口服给药的液相制剂有悬浮剂、内用液剂、乳剂、糖浆剂等,除了通常使用的作为简单稀释剂的水、液体石蜡之外,可包括多种赋形剂,如湿润剂、甜味剂、芳香剂、保鲜剂等。
用于非口服给药的制剂包括灭菌水溶液、非水溶剂、悬浮剂、乳剂、冷冻干燥剂、栓剂。作为非水溶剂、悬浮溶剂可使用丙二醇(propyleneglycol)、聚乙二醇、橄榄油等植物性油、油酸乙酯等可注射的酯等。作为栓剂的基质可使用witepsol、聚乙二醇、吐温(tween)61、可可油脂、月桂酸脂、甘油明胶等。
上述药学组合物可具有选自由注射剂、片剂、丸剂、散剂、颗粒剂、胶囊剂、悬浮剂、内用液剂、乳剂、糖浆剂、灭菌水溶液、非水溶剂、悬浮剂、乳剂、冷冻干燥剂以及栓剂组成的组中的一种剂型。
为了静脉内、皮肤或皮下注射等,活性成分可以为无热原(pyrogen-free)的具有适当ph、等渗性以及稳定性的用于非口服给药的、可接受的水溶液的形态。本技术领域的技术人员可通过使用诸如氯化钠水溶液、点滴液、乳酸点滴液等等渗性非载体来制备适当的溶液,在作为保鲜剂、稳定化剂、缓冲剂、抗氧化剂或其他不同的添加剂来需要的情况下可包含上述溶液。适合于注射的固体形态也可制备成对乳液或脂质体进行胶囊化的多肽的形态。
在本说明书中所使用的语句“有效量”或治疗有效量是指用于达成目的治疗结果(对于给药量及给药时间及方式)所需的量。有效量是至少赋予对象体治疗益处所需的活性剂的最小量,小于毒性量。例如,给药量范围为每位患者约100ng/kg至约100mg/kg,更典型地能够以约1μg/kg至约10mg/kg的范围给药。在活性化合物为盐、酯、酰胺、前体药物等的情况下,给药量以某化合物为基准计算,因此所使用的实际重量按比例增加。本发明的吡咯并苯并二氮杂化合物能够以每单位剂型(dosageform)包含0.1mg至3000mg、1mg至2000mg、10mg至1000mg的活性成分的方式被剂型化,但并不限定于此。可给药活性成分,以获得约0.05μm至100μm、1μm至50μm、5μm至30μm的活性化合物的峰等离子浓度。例如,任选地,在食盐水中通过静脉内注射0.1w/v%至5w/v%的活性成分的溶液来进行给药。
在药学组合物中,活性化合物的浓度可通过药物的吸收、非活性化、排出率以及本技术领域的普通技术人员已知的其他因素来确定。给药量可根据症状或疾病的严重程度而不同。并且,对于某一特定患者的给药量及给药方法可通过综合考虑患者的症状或疾病的程度、必要性、年龄、对药物的反应性等并根据给药监督人的职业判断来调整,在本发明中提出的浓度范围仅是作为一例,所请求保护的组合物的实施方式并不限定于此。并且,活性成分可以给药一次,或者可以分数次以较小给药量给药。
本发明的前体化合物或前体-连接体化合物、前体-连接体-配体缀合化合物可用于治疗增殖性疾病,尤其癌疾病。术语“增殖性疾病”是指无论是在试管内或生物体内,不合需要的过度或异常的细胞不会受到不希望的控制的细胞增殖相关疾病,如新生物或过形成性生长。例如,增殖性疾病包括新生物,肿瘤、癌、白血病、干癣、骨病、纤维化增生性疾病、动脉粥样硬化等,可包括阳性、所有恶性或恶性细胞增殖,但并不限定于此。上述癌可以为肺癌、小细胞肺癌、胃肠癌、大肠癌、肠癌、乳腺癌、卵巢癌、前列腺癌、睾丸癌、肝癌、肾癌、膀胱癌、胰腺癌、脑癌、肉瘤、骨肉瘤、卡波西肉瘤或黑色素瘤,但并不限定于此。
在本说明书中,除非另有定义,结合本发明所使用的科学术语及专业术语具有属于本技术领域的普通技术人员通常所理解的含义。
在本发明的一实施方式中,本发明的吡咯并苯并二氮杂前体、吡咯并苯并二氮杂前体-连接体化合物以及吡咯并苯并二氮杂-连接体-配体缀合物可通过如下过程来合成。
吡咯并苯并二氮杂前体的合成途径
吡咯并苯并二氮杂前体-连接体及吡咯并苯并二氮杂前体-连接体-配体缀合物的合成途径
本发明的吡咯并苯并二氮杂前体-连接体化合物及吡咯并苯并二氮杂前体-连接体-配体缀合物可通过使用在本说明书中提供的技术并利用本技术领域的技术人员的知识来制备。
例如,在本说明书中包含pct/us2016/063564号及pct/us2016/063595号中所描述的有关连接体的全部内容作为参照,其中,即使没有描述,还可根据在本说明书中所引用的或本技术领域的普通技术人员公知的参考文献来制备。
发明的效果
本发明的吡咯并苯并二氮杂二聚物前体、吡咯并苯并二氮杂二聚物前体-连接体或吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物本身的稳定性及血浆内稳定性优秀,在毒性表达方面具有优点,可对诸如癌等增殖性疾病进行靶向化、特异性治疗、药效最大化以及副作用表达最小化,因此可在产业上利用。
附图说明
图1为例示性地示出本发明的第28个化合物的合成过程。
具体实施方式
以下,通过实施例进一步详细说明本发明。但是,下述实施例有助于理解本发明,本发明的发明保护范围并不限定于此。
实施例1.化合物4的制备
化合物2的制备
将草酰氯(3.1ml,36.2mmol)溶解于二氯甲烷(40ml)后,在-78℃的温度、氮大气条件下,加入二甲基亚砜(4.7ml,66.4mmol)。10分钟后,缓慢加入将化合物1(10g,30.2mmol,化合物1利用j.org.chem.,2003,68,3923-3931中描述的方法制备)溶解于二氯甲烷(140ml)的溶液,将反应溶液搅拌1小时后加入三乙胺(16.7ml,120.6mmol),经2小时将反应温度逐渐升温至0℃。利用二氯甲烷(200ml)稀释反应溶液,利用饱和氯化铵水溶液(200ml)和盐水(200ml)洗涤有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物2(9.5g,95%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ4.39-4.26(m,1h),4.03-3.80(m,2h),3.69-3.64(m,1h),3.63-3.51(m,1h),2.70-2.60(m,1h),2.43(d,j=17.6hz),1.61-1.41(m,10h),0.98-0.67(m,6h),0.08-0.05(s,6h)。
化合物3的制备
利用四氢呋喃(80ml)稀释甲基三苯基溴化膦(7.6g,21.2mmol)后,在0℃的温度、氮大气条件下,加入叔丁醇钾(1minthf,21.2ml,21.2mmol)。搅拌1小时后,缓慢加入将化合物2(5.0g,15.2mmol)溶解于四氢呋喃(10ml)的溶液。将反应温度逐渐升温至常温并搅拌4小时。将饱和氯化铵水溶液(200ml)加入反应溶液后,利用二乙醚(2×200ml)提取。利用盐水(200ml)洗涤合并的有机层后,用无水硫酸钠干燥,过滤浓缩,经柱层析纯化得到化合物3(4.27g,86%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ4.97-4.91(m,2h),4.09-3.93(m,2h),3.84-3.80(m,1h),3.65-3.61(m,1h),3.59-3.34(m,1h),2.64-2.55(m,2h),1.69(s,9h),0.87(s,9h),0.03(s,6h)。
化合物4的制备
将化合物3(15.5g,47.2mmol)溶解于二氯甲烷(120ml)后,在0℃的温度下,加入盐酸(4n的1,4-二恶烷溶液,82.6ml,330.4mmol),在氮大气条件下,搅拌2小时。减压浓缩反应溶液获得白色固体化合物4(6.53g,92%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ9.79(brs,1h),9.17(brs,1h),5.15(d,j=8hz,1h),4.91(brs,1h),4.10(m,5h),2.76-2.70(m,1h),2.60-2.54(m,1h)。
实施例2.化合物9的制备
化合物6的制备
将化合物5(10g,20.2mmol,化合物5利用j.med.chem.,2004,47,1161-1174中描述的方法制备)溶解于二氯甲烷(100ml)后,在0℃的温度、氮大气条件下,加入草酰氯(6.1ml,70.8mmol)和n,n-二甲基甲酰胺(2滴)。将反应溶液搅拌4小时后,将温度升温至常温并搅拌10小时,减压浓缩后进行真空干燥。将获得的化合物溶解于二氯甲烷(120ml)后,在0℃的温度、氮大气条件下,加入化合物4(6.2g,41.4mmol)和三乙胺(9.9ml,70.8mmol)。将反应温度升温至常温并搅拌3小时后,将饱和氯化铵水溶液(200ml)加入反应溶液并利用二氯甲烷(2×200ml)提取。利用盐水(200ml)洗涤合并的有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物6(12g,87%)。
1h-nmr(400mhz,cdcl3)δ7.71(s,2h),6.80(s,2h),5.13(s,2h),4.88(s,2h),4.61(m,2h),4.17-4.14(t,j=6.2hz,4h),3.98(s,6h),3.94-3.74(m,10h),2.89-2.83(m,2h),2.52-2.48(m,2h),2.04-1.96(m,4h),1.77-1.71(m,2h)。
化合物7的制备
将化合物6(6.4g,9.36mmol)溶解于二氯甲烷(100ml)后,在0℃的温度、氮大气条件下,加入咪唑(2.5g,37.4mmol)和叔丁基二甲基氯硅烷(3.5g,23.4mmol)。将反应溶液搅拌2小时后,将饱和氯化铵水溶液(100ml)加入反应溶液并用二氯甲烷(2×100ml)提取。利用盐水(200ml)洗涤合并的有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物7(6.88g,75%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.70(s,1h),6.76(s,2h),4.99(s,2h),4.83(s,2h),4.59(brs,2h),4.14,(t,4h),3.95(s,6h),3.90(d,2h),3.77-3.69(m,4h),3.57(q,j=6.2hz,1h),3.31-3.29(m,1h),2.82-2.67(m,4h),1.99(t,j=7.2hz,4h),1.75-1.72(m,2h),0.89(s,18h),0.09(s,12h)。
化合物8的制备
将化合物7(3.0g,3.29mmol)溶解于乙醇(44ml)后,加入锌粉末(zincdust,12.9g,197mmol)和甲酸(5%的乙醇溶液,128ml)。在常温条件下,将反应溶液搅拌15分钟后,利用氟镁石过滤并加入乙酸乙酯(500ml)。依次用蒸馏水(200ml)、饱和碳酸氢钠水溶液(200ml)和盐水(200ml)洗涤有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物8(2.76g,98%)。
1h-nmr(400mhz,cdcl3)δ6.74(s,2h),6.24(s,2h),4.97(s,2h),4.90(s,2h),4.54(brs,2h),4.33(brs,4h),4.18(brs,1h),4.14(brs,2h),4.14-4.09(m,2h),4.00(t,j=8hz,4h),3.77(s,6h),3.62(brs.2h),2.68(s,4h),1.95-1.88(m,4h),1.66-1.64(m,2h),0.87(s,18h),0.02(s,12h)。
化合物9的制备
将化合物8(5.0g,5.86mmol)溶解于二氯甲烷(300ml)后,在-78℃的温度、氮大气条件下,加入吡啶(0.94ml,11.7mmol)和氯甲酸丙烯酯(0.62ml,5.86mmol)。将反应溶液搅拌1小时后,将温度升温至常温,浓缩后经柱层析纯化得到化合物9(2.23g,41%)。
1h-nmr(400mhz,cdcl3)δ7.84(s,1h),6.81(s,1h),6.74(s,1h),6.24(s,1h),5.98-5.92(m,1h),5.37,(dd,j=17.6hz,j=1.2hz,1h),5.25(dd,j=10.4hz,j=1.2hz,1h),4.97(brs,2h),4.90(brs,2h),4.63-4.62(m,4h),4.34(brs,2h),4.21-4.18(m,2h),4.10(t,j=6.4hz,3h),3.99(t,j=6.4hz,3h),3.83(s,3h),3.77(s,3h),3.63(bs,1h),2.68(brs,4h),1.97-1.89(m,4h),1.69-1.61(m,2h),0.87(s,18h),0.02(brs,12h)。
实施例3.化合物12的制备
化合物11的制备
将4-羟基苯甲醛(475mg,3.89mmol)和化合物10(1.7g,4.28mmol,化合物10利用韩国授权专利1628872中描述的方法制备)溶解于乙腈(40ml)后,加入
1h-nmr(400mhz,cdcl3)δ9.93(s,1h),7.86(d,j=8hz,2h),7.11(d,j=8.4hz,2h),5.38-5.29(m,4h),4.25-4.23(m,1h),3.71(s,3h),2.06(s,9h)。
化合物12的制备
将化合物11(1.3g,2.96mmol)溶解于氯仿/异丙醇(50ml/10ml)后,在0℃的温度、氮大气条件下,加入硅胶(1.3g)和氢化硼钠(134mg,3.55mmol)后搅拌2小时。将蒸馏水(40ml)加入反应溶液后,用乙酸乙酯(2×50ml)提取。将所提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物12(600mg,45%)。
1h-nmr(400mhz,cdcl3)δ7.31(d,j=8.4hz,2h),6.99(d,j=8.4hz2h),5.35-5.26(m,3h),5.13(d,j=7.6hz,1h),4.64(d,j=5.6hz,2h),4.18-4.16(m,1h),3.73(s,3h),2.06-2.04(m,9h),1.61(t,j=5.6hz,1h)。
实施例4.化合物15的制备
化合物13的制备
将5-甲酰水杨酸(5.0g,30.1mmol)溶解于甲醇(50ml)后,加入浓硫酸(2ml)。将反应溶液加热回流24小时后,减压浓缩并用乙酸乙酯(100ml)稀释。依次用蒸馏水(100ml)、饱和碳酸氢钠水溶液(200ml)和盐水(200ml)洗涤有机层后,用无水硫酸钠干燥。过滤浓缩,真空干燥得到白色固体化合物13(4.62g,85%)。
1h-nmr(400mhz,cdcl3)δ11.36(s,1h),9.88(s,1h),8.38(d,j=2.4hz,1h),8.00(dd,j=8.4hz,j=2hz,1h),7.11(d,j=8.8hz,1h),4.01(s,3h)。
化合物14的制备
将化合物13(1.7g,9.38mmol)和化合物10(4.1g,10.3mmol)溶解于乙腈(50ml)后,加入
1h-nmr(400mhz,cdcl3)δ9.95(s,1h),8.29(d,j=2hz,1h),8.01(dd,j=8.4hz,j=2hz,1h),7.26(d,j=8.8hz,1h),5.42-5.30(m,4h),4.27(d,j=9.2hz,1h),3.89(s,3h),3.72(s,3h),2.08(s,3h),2.07(s,3h),2.06(s,3h)。
化合物15的制备
将化合物14(2.85g,5.74mmol)溶解于氯仿:异丙醇(50ml/10ml)后,在0℃的温度、氮大气条件下,加入硅胶(2.8g)和氢化硼钠(434mg,11.5mmol)后,搅拌2小时。将蒸馏水(40ml)加入反应溶液后,利用二氯甲烷(2×50ml)提取。所提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物15(1.42g,49%)。
实施例5.化合物20的制备
化合物16的制备
将5-甲酰水杨酸(10.0g,60.1mmol)稀释于四氢呋喃(30ml)后,在常温条件下,加入n,n-二异丙基乙胺(29.8ml,180mmol)和苄基溴(7.15ml,60.1mmol)。将反应溶液加热回流18小时后,将温度降温至常温,加入2n的盐酸水溶液(100ml)。利用乙酸乙酯(2×100ml)提取该混合溶液,并用无水硫酸钠干燥合并的有机层。过滤浓缩,经柱层析纯化得到化合物16(12.9g,83%)。
1h-nmr(400mhz,cdcl3)δ11.38(s,1h),9.86(s,1h),8.40(s,1h),8.01(d,j=8.8hz,1h),7.44(m,5h),7.12(d,j=8.0hz,1h),5.42(s,2h)。
化合物17的制备
将化合物16(5.0g,19.5mmol)和化合物10(8.5g,21.4mmol)溶解于乙腈(100ml)后,加入
1h-nmr(400mhz,cdcl3)δ9.94(s,1h),8.28(s,1h),8.02(d,j=8.8hz,1h),7.46-7.28(m,6h),5.41-5.32(m,6h),4.27(d,j=9.2hz,1h),3.71(s,3h),2.06-2.04(m,9h)。
化合物18的制备
将化合物17(3.10g,5.41mmol)溶解于氯仿/异丙醇(45ml/9ml)后,在0℃的温度、氮大气条件下,加入硅胶(3g)和氢化硼钠(0.41g,10.8mmol)后,搅拌2小时。将蒸馏水(100ml)加入反应溶液后,用乙酸乙酯(200ml)提取。利用无水硫酸镁干燥所提取的有机层,过滤及减压浓缩后,经柱层析纯化得到白色固体化合物18(2.73g,87%)。
1h-nmr(400mhz,cdcl3)δ7.74(s,1h),7.48-7.34(m,6h),7.16(d,j=8.8hz,1h),5.35-5.26(m,5h),5.16-5.14(m,1h),4.17-4.15(m,1h),3.73(s,3h),2.04(s,9h),1.73(t,j=7.2hz,1h)。
化合物19的制备
将化合物18(2.40g,4.17mmol)溶解于乙醇(150ml)后,加入雷尼镍(raneyni,240mg)。在常温、氢大气条件下,将反应溶液搅拌10分钟。用氟镁石过滤反应溶液得到白色固体化合物19(2.10g)。
1h-nmr(400mhz,cdcl3)δ8.06(s,1h)7.61(d,j=8.8hz,1h),7.23(d,j=8.0hz1h),5.43-5.29(m,5h),4.17(s,2h),4.32(d,j=8.4hz,1h),3.69(s,3h),2.11-2.08(m,9h),1.24(t,1h)。
化合物20的制备
将化合物19(7.0g,14.5mmol)和2-甲氧基乙胺(1.38ml,1.59mmol)溶解于n,n-二甲基甲酰胺(14ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(6.57g,17.3mmol)和n,n-二异丙基乙胺(5ml,28.9mmol)。在常温条件下,将反应溶液搅拌2小时后,将饱和氯化铵水溶液(100ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取。利用盐水(200ml)洗涤合并的有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物20(7.53g,96%)。
1h-nmr(400mhz,cdcl3)δ7.98(d,j=2hz,1h),7.49(brs,1h),7.46(dd,j=8.4hz,j=2.4hz,1h),7.04(d,j=8.4hz,1h),5.42-5.28(m,4h),4.66(s,1h),4.19(d,j=9.2hz,1h),3.72(s,3h),3.57(s,3h),3.42(s,3h),2.05(s,9h)。
实施例6.化合物22的制备
将化合物19(1.0g,2.06mmol)和化合物21(1.49g,2.80mmol,化合物21利用pct/us2016/063564中描述的方法制备)溶解于n,n-二甲基甲酰胺(10ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(1.56g,4.12mmol)和n,n-二异丙基乙胺(1.07ml,6.18mmol)。在常温条件下,将反应溶液搅拌12小时后,将饱和氯化铵水溶液(100ml)加入反应溶液并利用乙酸乙酯(2×100ml)提取。利用盐水(200ml)洗涤合并的有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物22(1.6g,80%)。
1h-nmr(400mhz,cdcl3)δ7.98(s,1h),7.46(dd,j=8.4hz,j=2.4hz,1h),7.41(brs,1h),7.04(d,j=8.4hz,1h),5.93-5.25(m,4h),4.67(d,j=5.2hz,2h),4.20(d,j=9.6hz,1h),4.08(t,j=4.8hz,2h),3.74(s,6h),3.72-3.49(m,22h),2.06(s,9h),1.53(s,18h)。
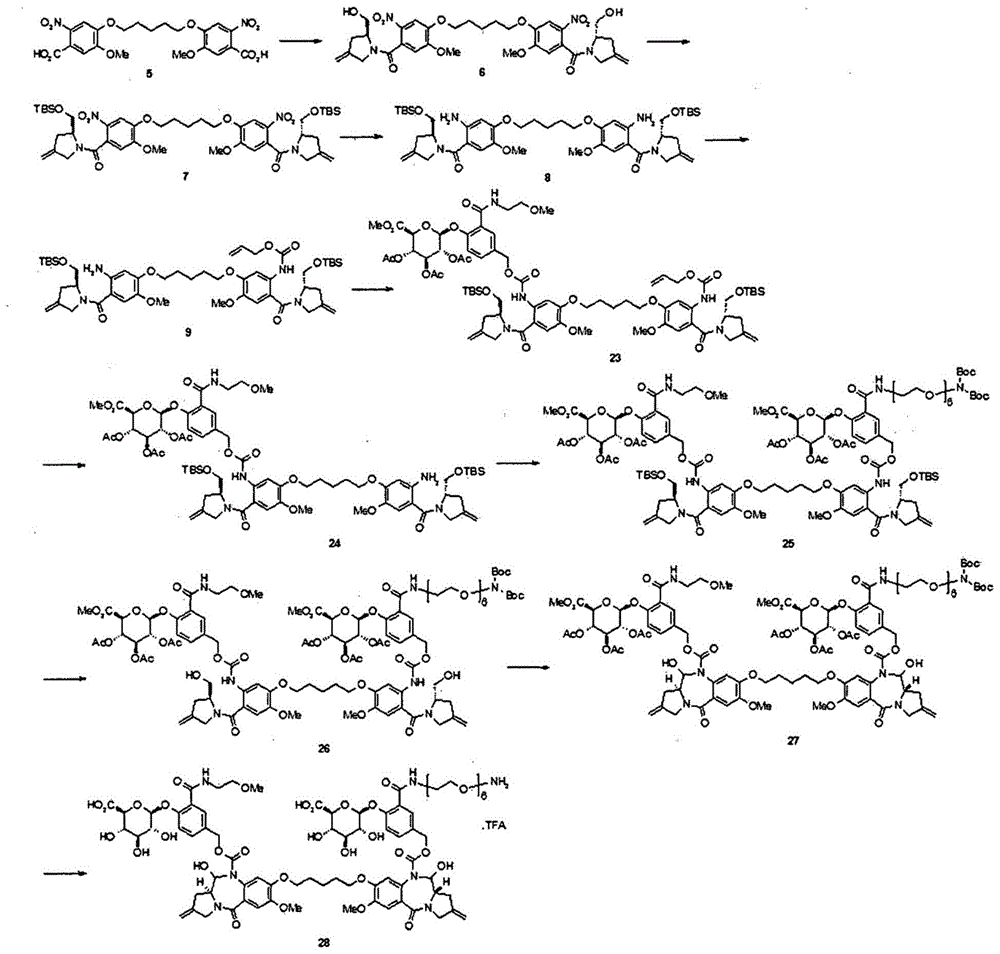
实施例7.化合物25的制备
化合物23的制备
将化合物9(2.2g,2.34mmol)溶解于甲苯(65ml)后,在-10℃的温度下,加入三光气(250mg,0.84mmol)和三乙胺(0.44ml,3.16mmol),在氮大气条件下,搅拌1小时。将化合物20(1.39g,2.58mmol)溶解于干燥的四氢呋喃(65ml),加入三乙胺(0.44ml,3.16mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(100ml)稀释后,用盐水(50ml)洗涤并用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物23(2.5g,72%)。
ei-msm/z:[m+h]+1504.7,1/2[m+h]+753.5。
化合物24的制备
将化合物23(2.0g,1.33mmol)溶解于二氯甲烷(15ml)后,加入吡咯啶(0.13ml,1.59mmol)和四(三苯基膦)钯(0)(76mg,0.066mmol),在常温、氮大气条件下,搅拌6小时。减压浓缩反应溶液后,经柱层析纯化得到化合物24(1.7g,90%)。
ei-msm/z:[m+h]+1420.6,1/2[m+h]+711.2。
化合物25的制备
将化合物24(1.2g,0.84mmol)溶解于甲苯(24ml)后,在-10℃的温度下,加入三光气(90mg,0.30mmol)和吡啶(0.33ml,4.22mmol),在氮大气条件下,搅拌1小时。将化合物22(974mg,1.01mmol)溶解于干燥的四氢呋喃(24ml),加入n,n-二异丙基乙胺(0.21ml,1.26mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(50ml)稀释后,利用盐水(30ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物25(800mg,40%)。
ei-msm/z:[m+h]+2409.9,1/2[m+na]+1214.3。
实施例8.化合物28的制备
化合物26的制备
将化合物25(800mg,0.33mmol)溶解于四氢呋喃/蒸馏水(4ml/4ml),加入乙酸(8ml)后,在常温、氮大气条件下,搅拌16小时。减压浓缩反应溶液后,经柱层析纯化得到化合物26(660mg,90%)。
ei-msm/z:[m+h]+2181.6,1/2[m-boc+h]+1041.5。
化合物27的制备
将化合物26(660mg,0.15mmol)溶解于二氯甲烷(15ml)后,加入戴斯-马丁试剂(dess-martinperiodinane,141mg,0.33mmol),在常温、氮大气条件下,搅拌3.5小时。减压浓缩反应溶液后,经柱层析纯化得到化合物27(477mg,70%)。
ei-msm/z:[m+h]+2177.6,1/2[m+h]+1089.5。
化合物28的制备
将化合物27(150mg,0.068mmol)溶解于甲醇/四氢呋喃(3ml/3ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(26mg,0.62mmol)溶解于蒸馏水(3ml)的溶液。将反应温度缓慢升温至0℃并搅拌2小时。用乙酸中和反应溶液后,减压浓缩并干燥反应溶液。利用二氯甲烷(5ml)稀释所得到的固体后,在0℃的温度下,加入三氟乙酸(1.2ml)并搅拌2小时。减压浓缩反应溶液后,经高效液相色谱(hplc)纯化,冷冻干燥得到白色固体化合物28(20mg,16%)。
ei-msm/z:[m+h]+1697.5,1/2[m+h]+849.3。
实施例9.化合物29的制备
化合物29利用化合物9和化合物12并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1596.9,1/2[m+h]+799.3。
实施例10.化合物30的制备
化合物30利用化合物9和化合物15并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1655.3,1/2[m+h]+828.1。
实施例11.化合物31的制备
化合物32利用化合物19和化合物31(化合物31利用pct/us2016/063564中描述的方法制备)并通过类似于化合物22的合成的方法制备。1h-nmr(400mhz,cdcl3)δ7.97(s,1h),7.46(dd,j=8.4hz,j=2.4hz,1h),7.41(bs,1h),7.04(d,j=8.4hz,1h),5.72(s,1h),5.42-5.27(m,4h),4.66(d,j=5.2hz,2h),4.25(d,j=9.6hz,1h),3.97(t,j=4.8hz,2h),3.78(s,3h),3.74-3.64(m,10h),2.04(s,9h),1.53(s,9h).ei-msm/z:[m+h]+731.5。
实施例12.化合物34的制备
化合物34利用化合物24和化合物32并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1565.5,1/2[m+h]+783.4.
实施例13.化合物39的制备
化合物35和化合物36利用pct/us2016/063564中描述的方法制备。
化合物37的制备
将化合物24(400mg,0.28mmol)溶解于甲苯(10ml)后,在-10℃的温度下,加入三光气(30mg,0.10mmol)和三乙胺(0.053ml,0.38mmol),在氮大气条件下,搅拌1小时。将化合物35(177mg,0.33mmol)溶解于干燥的四氢呋喃(10ml),加入三乙胺(0.053ml,0.38mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(50ml)稀释后,用盐水(30ml)洗涤并用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物37(192mg,34%)。
ei-msm/z:[m+h]+1971.8,1/2[m+h]+986.6。
化合物38的制备
将化合物37(192mg,0.097mmol)溶解于二氯甲烷(5ml)后,加入吡咯啶(0.012ml,0.14mmol)和四(三苯基膦)钯(0)(11.2mg,0.096mmol),在常温、氮大气条件下,搅拌6小时。减压浓缩反应溶液后,经柱层析纯化得到化合物38(180mg,96%)。
ei-msm/z:[m+h]+1932.8,1/2[m+h]+966.5。
化合物39的制备
将化合物38(180mg,0.093mmol)和化合物36(112mg,0.116mmol)溶解于n,n-二甲基甲酰胺(2ml)后,在0℃的温度、氮大气条件下,加入1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶-3-氧化物六氟磷酸盐(hatu,46mg,0.121mmol)和n,n-二异丙基乙胺(0.032ml,0.186mmol)。在常温条件下,将反应溶解搅拌36小时后,将蒸馏水(20ml)加入反应溶液,利用乙酸乙酯(2×20ml)提取。利用盐水(20ml)洗涤合并的有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物39(133mg,50%)。
ei-msm/z:[m+h]+2876.4,1/2[m+h]+1438.6。
实施例14.化合物42的制备
化合物40的制备
将化合物39(133mg,0.046mmol)溶解于四氢呋喃/蒸馏水(1ml/1ml),加入乙酸(2ml)后,在常温、氮大气条件下,搅拌16小时。减压浓缩反应溶液后,经柱层析纯化得到化合物40(67.4mg,55%)。
ei-msm/z:[m+h]+2647.4,1/2[m+h]+1324.5。
化合物41的制备
将化合物40(67.4mg,0.025mmol)溶解于二氯甲烷(2ml)后,加入戴斯-马丁试剂(dess-martinperiodinane,23.7mg,0.056mmol),在常温、氮大气条件下,搅拌3.5小时。减压浓缩反应溶液后,经柱层析纯化得到化合物41(43mg,65%)。
ei-msm/z:[m+h]+2643.1,1/2[m+h]+1322.5。
化合物42的制备
将化合物41(43mg,0.016mmol)溶解于甲醇/四氢呋喃(0.5ml/0.5ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(6.8mg,0.16mmol)溶解于蒸馏水(0.5ml)的溶液。将反应温度逐渐升温至-10℃并搅拌2小时。用乙酸中和反应溶液后,减压浓缩并干燥反应溶液。利用二氯甲烷(1ml)稀释所得到的固体后,在0℃的温度下,加入三氟乙酸(0.2ml)并搅拌2小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物42(7.0mg)。
ei-msm/z:[m+h]+2263.4,1/2[m+h]+1132.3。
实施例15.化合物48的制备
化合物44的制备
将化合物43(37g,40.2mmol,化合物43利用j.med.chem.,2004,47,1161-1174中描述的方法制备)溶解于二氯甲烷(400ml)后,在0℃的温度下,加入三氯异氰尿酸(14.9g,64.3mmol)和2,2,6,6-四甲基-1-哌啶酮(1.3g,8.0mmol),在氮大气条件下,搅拌1小时。向反应溶液加入二氯甲烷(400ml)来稀释,依次用饱和碳酸氢钠水溶液(400ml)、硫代硫酸钠(0.2m,400ml)水溶液和盐水(200ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物44(35g,83%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.72(s,2h),6.73(s,2h),4.97(d,2h),4.31(d,2h),4.12(t,4h),3.95-3.96(m,6h),3.71(d,2h),3.64(d,2h),3.45(d,2h),2.82-2.75(m,2h),2.55(d,2h),1.99(m,4h),1.72(m,2h),0.85(s,18h),0.08(d,12h)。
化合物45的制备
将化合物44(5g,5.45mmol)溶解于二氯甲烷(90ml)后,在-40℃的温度下,加入2,6-二甲基吡啶(5.1ml,43.8mmol)和三氟甲磺酸酐(5.5ml,39.0mmol),在氮大气条件下,搅拌1小时。向反应溶液加入二氯甲烷(90ml)来稀释,依次用饱和碳酸氢钠水溶液(90ml)、蒸馏水(90ml)和盐水(90ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物45(4.0g,62%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.71(s,2h),6.77(s,2h),6.08(s,2h),4.79-4.78(m,2h),4.18-4.09(m,6h),4.02-3.92(m,8h),3.22-3.14(m,2h),3.01-2.97(m,2h),2.02-1.97(m,4h),0.91(s,18h),0.11(s,12h)。
化合物46的制备
将化合物45(3.1g,2.6mmol)溶解于甲苯(45ml)后,在氩大气条件下,加入甲基硼酸(1.1g,18.2mmol)、氧化银(i)(4.8g,20.9mmol)、磷酸钾(6.6g,31.5mmol)、三苯胂(642mg,2.1mmol)和双(三苯基膦)二氯化钯(ii)(184mg,0.3mmol),在80℃的温度下,加热搅拌3小时。利用氟镁石过滤反应溶液后,浓缩,经柱层析纯化得到化合物46(955mg,40%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.68(s,2h),6.77(s,2h),5.52(s,2h),4.66-4.64(m,2h),4.14-4.07(m,6h),3.94-3.92(m,8h),2.75-2.73(m,2h),2.55-2.51(m,2h),1.99-1.93(m,4h),1.72-1.68(m,2h),1.60(s,6h),0.88(s,18h),0.09(s,12h)。
化合物47的制备
将化合物46(2.9g,3.17mmol)溶解于乙醇(44ml)后,加入锌粉末(zincdust,12.9g,197mmol)和甲酸(5%的乙醇溶液,128ml)。在常温条件下,将反应溶液搅拌15分钟后,利用氟镁石过滤,并加入乙酸乙酯(500ml)。依次用蒸馏水(200ml)、饱和碳酸氢钠水溶液(200ml)和盐水(200ml)洗涤有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物47(3.0g,82%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ6.74(s,2h),6.23(s,2h),6.18(bs,2h),4.64(bs,2h),4.34(s,3h),4.07-3.93(m,6h),3.80-3.76(m,7h),2.74-2.68(m,2h),2.53(d,2h),1.91(m,4h),1.67-1.62(m,8h),0.88(s,18h),0.05(d,12h)。
化合物48的制备
将化合物47(3.0g,3.51mmol)溶解于二氯甲烷(175ml)后,在-78℃的温度、氮大气条件下,加入吡啶(0.57ml,7.03mmol)和氯甲酸丙烯酯(0.34ml,3.16mmol)。将反应溶液搅拌1小时后,将反应温度升温至常温,浓缩后,经柱层析纯化得到化合物48(1.33g,44%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ8.80(brs,1h),7.82(s,1h),6.78(s,1h),6.74(s,1h),6.23(s,1h),6.19(brs,2h),5.99-5.90(m,1h),5.34(d,1h),5.23(d,1h),4.63(m,4h),4.35(brs,2h),4.10(t,2h),3.99(t,3h),3.99(m,2h),3.80(s,5h),3.76(s,4h),2.73(m,2h),2.55(m,2h),1.95-1.90(m,4h),1.68-1.63(m,8h),0.88(s,18h),0.05(d,12h)。
实施例16.化合物49的制备
将化合物19(3.7g,7.56mmol)和丙炔胺(0.43ml,7.07mmol)溶解于n,n-二甲基甲酰胺(50ml)后,加入n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(2.32g,12.1mmol)和1-羟基苯并三唑(2.04g15.1mmol)。在常温条件下,将反应溶液搅拌12小时后,将蒸馏水(100ml)加入反应溶液,用乙酸乙酯(2×100ml)提取。利用盐水(200ml)洗涤合并的有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物49(3.4g,86%)。
1h-nmr(400mhz,cdcl3)δ8.01(d,1h),7.57(t,1h),7.49(dd,1h),7.02(d,1h),5.42-5.38(m,1h),5.36-5.28(m,2h),4.67(d,2h),4.31-4.13(m,3h),2.23(t,1h),2.07-2.06(m,9h),1.88(t,1h)。
实施例17.化合物53的制备
化合物51的制备
将化合物50(4.5g,25.68mmol)溶解于n,n-二甲基甲酰胺(50ml)后,在0℃的温度、氮大气条件下,加入氢化钠(1.23g,30.82mmol)搅拌30分钟后,加入溴丙炔(~80%的甲苯溶液,4.96ml,33.4mmol)后,在常温条件下,搅拌3小时。将蒸馏水(40ml)加入反应溶液后,用乙酸乙酯(2×50ml)提取。利用盐水(100ml)洗涤合并的有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物51(4.35g,79%)。
1h-nmr(400mhz,cdcl3)δ4.21(d,j=2.4hz,2h),3.70-3.38(m,10h),3.39(t,j=5.2hz,2h),2.43(t,j=2.4hz,1h)。
化合物52的制备
将化合物51(1.55g,7.03mmol)溶解于干燥的四氢呋喃(30ml)/蒸馏水(2.53ml)后,加入三苯基膦(2.21g,8.44mmol),在常温条件下,搅拌24小时。浓缩,经柱层析纯化得到化合物52(1.3g,99%)。
1h-nmr(400mhz,cdcl3)δ4.21(s,2h),3.69-3.64(m,8h),3.52-3.49(m,2h),2.88-2.85(m,2h),4.23(s,1h).ei-msm/z:[m+h]+188.2。
化合物53的制备
将化合物52(2.0g,10.68mmol)和化合物19(4.7g,9.71mmol)溶解于n,n-二甲基甲酰胺(50ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(3.71g,11.6mmol)和n,n-二异丙基乙胺(3.38ml,19.4mmol)。在常温条件下,将反应溶液搅拌24小时后,将蒸馏水(100ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取。利用盐水(200ml)洗涤合并的有机层后用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物53(4.78g,75%)。
1h-nmr(400mhz,cdcl3)δ7.95(s,1h),7.46(d,j=8.4hz,1h),7.41-7.37(m,1h),7.04(d,j=8.8hz,1h),5.41-5.25(m,4h),4.65(d,j=4.4hz,2h),4.21(d,j=9.2hz,1h),4.17(s,2h),3.74(s,3h),3.68(s,11h),3.56-3.50(m,1h),2.05(s,9h)。
实施例18.化合物55的制备
化合物55的制备
将化合物19(3.68g,7.60mmol)和化合物54(1.46g,8.40mmol,化合物54利用pct/us2016/063564中描述的方法制备)溶解于n,n-二甲基甲酰胺(10ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(4.53g,11.40mmol)和n,n-二异丙基乙胺(3.97ml,22.80mmol)后,在常温条件下,搅拌12小时。将饱和氯化铵水溶液(100ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取后,利用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物55(3.31g,68%)。
1h-nmr(400mhz,cdcl3)δ7.99(s,1h),7.47(d,j=8.4hz,1h),7.41(s,1h),5.42-5.25(m,4h),4.68(d,j=5.6hz,2h),4.20(d,j=9.2hz,1h),3.78-3.68(m,11h),3.58-3.52(m,1h),3.39-3.36(m,2h),2.06(s,9h),1.89-1.86(m,1h)。
实施例19.化合物58的制备
化合物56的制备
将化合物48(1.33g,1.41mmol)溶解于甲苯(40ml)后,在-10℃的温度下,加入三光气(151mg,0.51mmol)和三乙胺(0.26ml,1.91mmol),在氮大气条件下,搅拌1小时。将化合物20(845mg,1.56mmol)溶解于干燥的四氢呋喃(40ml),加入三乙胺(0.26ml,1.91mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(30ml)稀释后,用盐水(20ml)洗涤并用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物56(1.15mg,54%)。
ei-msm/z:[m+h]+1504.7,1/2[m+h]+753.5。
化合物57的制备
将化合物56(1.15g,0.79mmol)溶解于二氯甲烷(10ml)后,加入吡咯啶(0.08ml,1.35mmol)和四(三苯基膦)钯(0)(45mg,0.057mmol),在常温、氮大气条件下,搅拌2小时。减压浓缩反应溶液后,经柱层析纯化得到化合物57(820mg,72%)。
ei-msm/z:[m+h]+1420.6,1/2[m+h]+711.2。
化合物58的制备
将化合物57(730mg,0.51mmol)溶解于甲苯(20ml)后,在-10℃的温度下,加入三光气(54mg,0.36mmol)和吡啶(0.2ml,2.56mmol),在氮大气条件下,搅拌1小时。将化合物49(321mg,0.61mmol)溶解于干燥的四氢呋喃(20ml),加入n,n-二异丙基乙胺(0.14ml,0.77mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(50ml)稀释后,用盐水(30ml)洗涤并用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物58(650mg,64%)。
ei-msm/z:[m+h]+1969.2,1/2[m+h]+985.2。
实施例20.化合物61的制备
化合物59的制备
将化合物58(650mg,0.33mmol)溶解于四氢呋喃/蒸馏水(3.5ml/3.5ml),加入乙酸(7ml)后,在常温、氮大气条件下,搅拌16小时。减压浓缩反应溶液后,经柱层析纯化得到化合物59(440mg,78%)。
ei-msm/z:[m+h]+1740.0,[m+na]+1762.0,1/2[m+h]+871.0。
化合物60的制备
将化合物59(440mg,0.25mmol)溶解于二氯甲烷(25ml)后,加入戴斯-马丁试剂(dess-martinperiodinane,236mg,0.55mmol),在常温、氮大气条件下,搅拌2.5小时。减压浓缩反应溶液后,经柱层析纯化得到化合物60(365mg,84%)。
ei-msm/z:[m+h]+1736.0,1/2[m+h]+869.5。
化合物61的制备
将化合物60(365mg,0.21mmol)溶解于甲醇/四氢呋喃(9ml/2ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(58mg,1.4mmol)溶解于蒸馏水(9ml)的溶液。将反应温度缓慢升温至0℃并搅拌2小时。用乙酸中和反应溶液后,减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物61(100mg,32%)。
ei-msm/z:[m+h]+1456.8,1/2[m+h]+729.5。
实施例21.化合物62的制备
化合物62利用化合物53和化合物57并通过类似于化合物61的合成的方法制备。ei-msm/z:[m+h]+1588.7,1/2[m+h]+795.3。
实施例22.化合物63的制备
化合物63利用化合物55和化合物57并通过类似于化合物61的合成的方法制备。ei-msm/z:[m+h]+1575.8,1/2[m+h]+788.7。
实施例23.化合物65的制备
化合物65的制备
将化合物61(100mg,0.068mmol)溶解于二甲基亚砜(1.6ml)后,在氮大气条件下,加入化合物64(136mg,0.302mmol,化合物64利用pct/us2016/063564中描述的方法制备)后,在蒸馏水(0.4ml)中溶解五水合硫酸铜(ii)(7.4mg,0.03mml)和抗坏血酸钠(28mg,0.15mmol)来加入反应溶液中。在常温条件下,搅拌30分钟,减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物65(15.2mg,13%)。
ei-msm/z:[m+h]+1645.8,1/2[m+h]+823.9。
实施例24.化合物70的制备
化合物66的制备
将3-氨基-1-丙醇(3.0g,66.57mmol)溶解于二氯甲烷(150ml)后,在0℃的温度、氮大气条件下,加入二碳酸二叔丁酯(16g,73.2mmol)。在常温条件下,搅拌12小时后,减压浓缩反应溶液,经柱层析纯化得到化合物66(6.4g,92%)。
1h-nmr(400mhz,cdcl3)δ4.78(s,1h),3.65(m,2h),3.30(m,2h),2.90(s,1h),1.68(m,2h),1.48(s,9h)。
化合物67的制备
将化合物66(6.04g,34.47mmol)和三乙胺(14.4ml,103.4mmol)溶解于四氢呋喃(100ml)后,在0℃的温度、氮大气条件下,缓慢加入甲磺酸酐(7.21g,41.36mmol)。逐渐升温至室温后,搅拌12小时。减压浓缩反应溶液,经柱层析纯化得到化合物67(9.01g,98%)。
1h-nmr(400mhz,cdcl3)δ4.73(s,1h),4.30(t,j=5.9hz,2h),3.31-3.24(m,2h),3.04(s,3h),1.94(t,j=6.1hz,2h),1.44(s,9h)。
化合物68的制备
将化合物67(3.0g,11.84mol)溶解于n,n-二甲基甲酰胺(40ml)后,在常温、氮大气条件下,加入叠氮化钠(924mg,14.21mmol),在60℃的温度下,将反应混合物搅拌12小时。将蒸馏水(50ml)和1n的盐酸水溶液(5ml)加入反应溶液,利用乙酸乙酯(100ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物68(2.3g,99%)。
1h-nmr(600mhz,cdcl3)δ4.63(s,1h),3.36(t,j=6.6hz,2h),3.24-3.18(m,2h),1.80-1.75(m,2h),1.45(s,9h)。
化合物69的制备
将化合物68(3.8g,18.98mmol)溶解于二氯甲烷(10ml),在0℃的温度、氮大气条件下,缓慢加入盐酸(4m的1,4-二恶烷溶液,10ml)。将反应混合物搅拌12小时后,通过减压浓缩得到化合物69(2.5g,99%)。
1h-nmr(600mhz,dmso-d6)δ8.06(s,3h),3.47(t,j=6.6hz,2h),2.82(t,j=7.2hz,2h),1.84-1.79(m,2h)。
化合物70的制备
将化合物19(4.1g,8.46mmol)和化合物69(1.1g,11.0mmol)溶解于n,n-二甲基甲酰胺(溶解于20ml),在0℃、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(4.39g,11.0mmol)和n,n-二异丙基乙胺(2.96ml,16.92mmol)后,在常温条件下,搅拌12小时。将饱和氯化铵水溶液(100ml)加入反应溶液并用乙酸乙酯(2×100ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物70(5.48g,88%)。
1h-nmr(400mhz,cdcl3)δ8.07(s,1h),7.50-7.46(m,2h),7.01(d,j=8.4hz,1h),5.45-5.30(m,4h),4.69(d,j=5.6hz,2h),4.21(d,j=9.6hz,1h),3.74(s,3h),3.67-3.60(m,1h),3.47-3.41(m,3h),2.80(s,2h),2.07-2.05(m,9h),1.98-1.91(m,2h),1.80-1.77(m,1h)。
实施例25.化合物72的制备
化合物72的制备
将化合物19(3.6g,7.42mmol)和化合物71(1.0g,8.16mmol,化合物71利用类似于化合物69的方法制备)溶解于n,n-二甲基甲酰胺(15ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(4.2g,11.2mmol)和n,n-二异丙基乙胺(3.2ml,18.6mmol)。在常温条件下,将反应溶液搅拌14小时后,将饱和氯化铵水溶液(50ml)加入反应溶液,用乙酸乙酯(2×50ml)提取。利用盐水(50ml)洗涤合并的有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物72(3.9g,95%)。
ei-msm/z:[m+h]+553.3,[m+na]+575.4。
实施例26.化合物73的制备
化合物73利用化合物24和化合物55并通过类似于化合物63的合成的方法制备。ei-msm/z:[m+h]+1575.7,1/2[m+h]+788.8。
实施例27.化合物74的制备
化合物74利用化合物24和化合物70并通过类似于化合物63的合成的方法制备。ei-msm/z:[m+h]+1500.9,1/2[m+h]+751.2。
实施例28.化合物75的制备
化合物75利用化合物24和化合物72并通过类似于化合物63的合成的方法制备。ei-msm/z:[m+h]+1486.42,[m+na]+1509.31。
实施例29.化合物80的制备
化合物77的制备
将化合物76(7.30g,28.5mmol,化合物76利用angew.chem.int.ed.,2016,55,12338-12342中描述的方法制备)和化合物16(14.0g,29.4mmol)溶解于乙腈(145ml)后,加入
1h-nmr(400mhz,cdcl3)δ9.94(s,1h),8.27(s,1h),7.99(d,j=8.8hz,1h),7.46-7.29(m,6h),5.64-5.59(m,1h),5.49-5.48(m,1h),5.36(s,2h),5.18(d,j=8.0hz,1h),5.19-5.11(m,1h),4.27-4.10(m,3h),2.19(s,3h),2.08(s,3h),2.04(s,3h),2.03(s,3h)。
化合物78的制备
将化合物77(15.30g,26.10mmol)溶解于氯仿/异丙醇(200ml/40ml)后,在0℃、氮大气条件下,加入硅胶(16g)和氢化硼钠(1.53g,40.50mmol)后,搅拌30分钟。将蒸馏水(200ml)加入反应溶液后,利用乙酸乙酯(400ml)提取。利用无水硫酸镁干燥所提取的有机层,经过滤及减压浓缩得到化合物78(14.0g,91%)。
1h-nmr(400mhz,cdcl3)δ7.73(s,1h),7.47-7.31(m,6h),7.19(d,j=8.4hz,1h),5.57(t,j=9.2hz,1h),5.46(d,j=3.2hz,1h),5.36-5.28(m,2h),5.12-5.04(m,2h),4.66(d,j=6.0hz,2h),4.26-4.04(m,3h),2.18(s,3h),2.07(s,3h),2.05(s,3h),2.02(s,3h),1.67(t,j=5.6hz,1h)。
化合物79的制备
将化合物78(14.0g,23.8mmol)溶解于乙醇(550ml)后,加入雷尼镍(raneyni,14.0g)。在常温、氢大气条件下,将反应溶液搅拌12小时。利用氟镁石过滤反应溶液,浓缩得到化合物79(11.4g,96%)。
1h-nmr(400mhz,cdcl3)δ8.07(s,1h)7.58(d,j=8.8hz,1h),7.17(d,j=8.8hz1h),5.57(t,j=9.2hz,1h),5.49(d,j=3.2hz,1h),5.22(d,j=8.0hz,1h),5.17-5.14(m,1h),4.71(s,2h),4.25-4.10(m,3h),2.20(s,3h),2.11(s,3h),2.07(s,3h),2.00(s,3h)。
化合物80的制备
将化合物79(3.00g,6.00mmol)和2-甲氧基乙胺(0.57ml,6.6mmol)溶解于n,n-二甲基甲酰胺(15ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(2.86g,7.20mmol)和n,n-二异丙基乙胺(2.10ml,12.0mmol)后,在常温条件下,搅拌12小时。将饱和氯化铵水溶液(100ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物80(2.3g,68%)。
1h-nmr(400mhz,cdcl3)δ8.07(s,1h),7.48-7.44(m,2h),7.06(d,j=8.4hz,1h),5.55(t,j=9.2hz,1h),5.49(d,j=2.8hz,1h),5.20-5.14(m,2h),4.69(d,j=5.2hz,2h),4.25-4.09(m,3h),3.78-3.74(m,1h),3.62-3.51(m,3h),3.40(s,3h),2.21(s,3h),2.07(m,6h),2.02(s,3h),1.71(t,j=6.0hz,1h)。
实施例30.化合物81的制备
化合物81的制备
将化合物79(2.19g,4.38mmol)和化合物31(1.50g,5.70mmol)溶解于n,n-二甲基甲酰胺(10ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(2.26g,5.7mmol)和n,n-二异丙基乙胺(1.53ml,8.76mmol)后,在常温条件下,搅拌12小时。将饱和氯化铵水溶液(100ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物81(2.73g,84%)。
1h-nmr(400mhz,cdcl3)δ8.07(s,1h),7.68(s,1h),7.46-7.44(m,2h),7.05(d,j=8.0hz,1h),5.56-5.49(m,2h),5.18-5.14(m,2h),4.68(d,j=4.8hz,2h),4.27-4.10(m,3h),3.97-3.95(m,2h),3.83-3.78(m,1h),3.71-3.67(m,7h),3.57-3.52(m,1h),2.22(s,3h),2.07(m,6h),2.03(s,3h),1.47(s,9h)。
实施例31.化合物82的制备
化合物82利用化合物9、化合物80和化合物81并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1537.7,1/2[m+h]+769.7。
实施例32.化合物83的制备
化合物83利用化合物9、化合物80和化合物32并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1551.6,1/2[m+h]+776.7。
实施例33.化合物85的制备
化合物85利用化合物9、化合物84(化合物84利用wo2011/130598a1中描述的方法制备)和化合物32并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1587.8,1/2[m+h]+794.7。
比较例1.化合物86、化合物87和化合物88的制备
化合物86、化合物87以及化合物88利用pct/us2016/063564中描述的方法制备。
实施例34.化合物94的制备
化合物90的制备
化合物89(4.5g,4.88mmol,化合物89利用j.med.chem.,2004,47,1161-1174中描述的方法制备)溶解于二氯甲烷(100ml)后,在常温、氮大气条件下,加入2,2,6,6-四甲基-1-哌啶酮(153mg,0.98mmol)和(二乙酰氧基碘)苯(7.0g,21.7mmol)。将反应溶液搅拌24小时后,将蒸馏水(200ml)加入反应溶液,利用二氯甲烷(2×200ml)提取。利用无水硫酸钠干燥合并的有机层,过滤,减压浓缩后,经柱层析纯化得到化合物90(4.25g,95%)。
1h-nmr(400mhz,cdcl3)δ7.74(s,2h),6.73(s,2h),4.97(d,j=8.8hz,1h),4.39-4.27(m,8h),3.96(s,6h),3.80-3.70(m,2h),3.58-3.52(m,2h),3.42-2.79(m,2h),2.74-2.56(m,2h),2.52-2.44(m,2h),2.08(s,2h),0.85(s,18h),0.97(s,12h)。
化合物91的制备
将化合物90(10.0g,10.9mmol)溶解于二氯甲烷(450ml)后,在-40℃的温度、氮大气条件下,加入2,6-二甲基吡啶(10.0ml,87.2mmol)和三氟甲磺酸酐(11.0ml,65.4mmol)。将反应溶液搅拌2小时后,将饱和碳酸氢钠水溶液(500ml)加入反应溶液,利用二氯甲烷(2×500ml)提取。利用无水硫酸钠干燥合并的有机层,过滤,减压浓缩后,经柱层析纯化得到化合物91(10.6g,47%)。
化合物92的制备
将化合物91(1.7g,1.44mmol)溶解于乙醇/甲苯/蒸馏水(12ml/24ml/12ml)后,在常温、氮大气条件下,加入4-甲基苯硼酸(568mg,3.74mmol)、碳酸钠(793mg,7.48mmol)以及四(三苯基膦)钯(0)(133mg,0.115mmol)。将反应溶液搅拌2小时后,利用乙酸乙酯(100ml)稀释反应溶液,利用盐水(100ml)和蒸馏水(100ml)清洗有机层。利用无水硫酸钠干燥合并的有机层,过滤,减压浓缩后,经柱层析纯化得到化合物92(1.25g,79%)。
1h-nmr(400mhz,cdcl3)δ7.80(s,2h),7.13(d,j=8.8hz,4h),6.90(s,2h),6.79(d,j=8.0hz,4h),6.14(s,2h),4.80-4.50(m,2h),4.39-4.36(m,4h),3.98(s,6h),3.79(s,6h),3.17(bs,2h),3.02-2.98(m,2h),2.50-2.47(m,2h),0.88(s,18h),0.11(s,12h).ei-msm/z:[m+h]+1069.8,1/2[m+h]+535.6。
化合物93的制备
将化合物92(8.0g,7.48mmol)溶解于乙醇(300ml)后,加入锌粉末(zincdust,29g,28.1mmol)和甲酸(5%,处于etoh中,320ml)。在常温条件下,将反应溶液搅拌20分钟后,利用氟镁石过滤并添加乙酸乙酯(1l)。依次用蒸馏水(500ml)、饱和碳酸氢钠水溶液(500ml)以及盐水(500ml)洗涤有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物93(4.85g,64%)。
1h-nmr(400mhz,cdcl3)δ7.16(d,j=8.4hz,4h),6.78(d,j=6.4hz,8h),6.30(s,2h),4.71-4.41(m,2h),4.25(brs,4h),4.19-4.17(m,4h),4.10-4.05(m,2h),3.95-3.81(m,2h),3.73(s,6h),3.72(s,6h),3.64-3.10(m,2h),3.03-2.93(m,2h),2.36-2.34(m,2h),0.81(s,18h),0.11(s,12h).ei-msm/z:[m+h]+1010.4,1/2[m+h]+505.7。
化合物94的制备
将化合物93(4.6g,4.56mmol)溶解于二氯甲烷(300ml)后,在-78℃的温度、氮大气条件下,加入吡啶(0.74ml,9.11mmol)和氯甲酸丙烯酯(0.48ml,4.56mmol)。将反应溶液搅拌1小时后,将反应温度升温至常温,浓缩后,经柱层析纯化得到化合物94(1.46g,29%)。
ei-msm/z:[m+h]+1093.6。
实施例35.化合物97的制备
化合物95的制备
将化合物94(200mg,0.18mmol)溶解于甲苯(7.5ml)后,在-10℃的温度下,加入三光气(19mg,0.067mmol)和三乙胺(0.035ml,0.25mmol),在氮大气条件下,搅拌1小时。将化合物20溶解于干燥的四氢呋喃(7.5ml),加入三乙胺(0.035ml,0.25mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,用二氯甲烷(30ml)稀释后,用盐水(20ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物95(130mg,43%)。
ei-msm/z:[m+h]+1661.6,1/2[m+h]+831.4。
化合物96的制备
将化合物95(380mg,0.23mmol)溶解于二氯甲烷(10ml)后,依次加入吡咯啶(0.023ml,0.27mmol)、四(三苯基膦)钯(0)(13mg,0.011mmol)以及三苯基膦(15mg,0.057mmol),在常温、氮大气条件下,搅拌6小时。减压浓缩反应溶液后,经柱层析纯化得到化合物96(260mg,72%)。
ei-msm/z:[m+h]+1577.6,1/2[m+h]+789.4。
化合物97的制备
将化合物96(260mg,0.16mmol)溶解于甲苯(5ml)后,在-10℃的温度下,加入三光气(17.6mg,0.06mmol)和二异丙基乙胺(0.053ml,0.30mmol),在氮大气条件下,搅拌1小时。将化合物22溶解于干燥的四氢呋喃(5ml),加入吡啶(0.066ml,0.80mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(50ml)稀释后,用盐水(30ml)洗涤并用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物97(168mg,41%)。
ei-msm/z:[m+h]+2567.1,1/2[m+h]+1283.8。
实施例36.化合物100的制备
化合物98的制备
将化合物97(168mg,0.065mmol)溶解于四氢呋喃/蒸馏水(1ml/1ml),加入乙酸(2ml)后,在常温、氮大气条件下,搅拌16小时。减压浓缩反应溶液后,经柱层析纯化得到化合物98(130mg,85%)。
ei-msm/z:[m+h]+2337.8,1/2[m+h]+1169.5。
化合物99的制备
将化合物98(130mg,0.055mmol)溶解于二氯甲烷(5ml)后,加入戴斯-马丁试剂(dess-martinperiodinane,57mg,0.13mmol),在常温、氮大气条件下,搅拌3.5小时。减压浓缩反应溶液后,经柱层析纯化得到化合物99(96mg,82%)。
ei-msm/z:[m+h]+2333.7,1/2[m+h]+1167.5。
化合物100的制备
将化合物99(96mg,0.041mmol)溶解于甲醇/四氢呋喃(1ml/1ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(16mg,0.41mmol)溶解于蒸馏水(1ml)的溶液。将反应温度缓慢升温至0℃并搅拌2小时。用乙酸中和反应溶液后,减压浓缩并干燥反应溶液。利用二氯甲烷(2ml)稀释所得到的固体后,在0℃的温度下,加入三氟乙酸(0.5ml)并搅拌2小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到淡绿色固体化合物100(2.4mg)。
ei-msm/z:[m+h]+1853.8,1/2[m+h]+927.4。
实施例37.化合物102的制备
化合物101的制备
将1,3-二氨基丙烷(0.93ml,11.1mmol)溶解于二氯甲烷(30ml),在0℃的温度、氮大气条件下,加入二碳酸二叔丁酯(0.84ml,3.7mmol)。在常温条件下,搅拌3小时后,将盐水(50ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取后,用无水硫酸钠干燥。过滤后,减压浓缩反应溶液,经柱层析纯化得到化合物101(658mg,以100%的boc2o为基准)。
1h-nmr(400mhz,cdcl3)δ4.88(brs,1h),3.26-3.14(m,2h),2.77(t,j=6.8hz,2h),1.66-1.57(m,2h),1.44(s,9h),1.32(br,2h)。
化合物102的制备
将化合物19(1.50g,3.10mmol)和化合物101(0.65g,3.73mmol)溶解于n,n-二甲基甲酰胺(10ml),在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(1.60g,4.03mmol)和n,n-二异丙基乙胺(1.08ml,6.20mmol)后,在常温条件下,搅拌12小时。将饱和氯化铵水溶液(100ml)加入反应溶液,利用乙酸乙酯(2×100ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物102(1.67g,84%)。
1h-nmr(400mhz,cdcl3)δ8.05(s,1h),7.49-7.47(m,2h),7.02(d,j=8.4hz,1h),5.42-5.30(m,4h),4.69(d,j=6.0hz,2h),4.21(d,j=9.2hz,1h),3.74(s,3h),3.63-3.58(m,1h),3.44-3.39(m,1h),322-3.13(m,2h),2.06-2.05(m,9h),1.79-1.74(m,2h),1.45(s,9h)。
实施例38.化合物103的制备
化合物103利用化合物24和化合物102并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1475.8,1/2[m+h]+738.3。
实施例39.化合物104的制备
将化合物103(35mg,0.024mmol)和马来酰亚胺基乙酸n-羟基琥珀酰亚胺酯(9mg,0.035mmol)溶解于n,n-二甲基甲酰胺(1.5ml)后,在0℃的温度、氮大气条件下,加入n,n-二异丙基乙胺(0.021ml,0.23mmol)。将反应温度逐渐升温至常温后,搅拌3小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物104(15.1mg,37%)。
ei-msm/z:[m+h]+1612.6,1/2[m+h]+807.2。
实施例40.化合物110的制备
化合物105的制备
将l-天冬酰胺(3.0g,22.7mmol)溶解于1n的碳酸钠水溶液(30ml)后,在0℃的温度下,加入氯甲酸苄酯(6.3ml,45.4mmol),在氮大气条件下,搅拌12小时。将蒸馏水(50ml)加入反应溶液后,利用1n的盐酸水溶液进行酸性化(ph2)。利用乙酸乙酯(3×50ml)提取该混合物,利用无水硫酸钠干燥合并的有机层。过滤后,浓缩获得化合物105(3.5g,58%)。
1h-nmr(400mhz,dmso-d6)δ7.51-7.40(d,j=8.0hz,1h),7.35(s,6h),6.92(s,1h),5.02(s,2h),2.61-2.35(m,2h)。
化合物106的制备
将化合物105(3.5g,13.1mmol)溶解于乙酸乙酯/乙腈/蒸馏水(30ml/30ml/15ml)后,加入(二乙酰氧基碘)苯(5.1g,15.7mmol),在氮大气条件下,搅拌10小时。通过过滤及减压浓缩所形成的固体来得到化合物106(2.8g,89%)。
1h-nmr(400mhz,dmso-d6)δ9.94(s,1h),7.96(s,2h),7.72(d,j=8.8hz,1h),7.37(s,5h),5.07(s,2h),4.29(s,1h),3.23(br,1h),3.02(br,1h)。
化合物107的制备
将化合物106(2.8g,11.7mmol)溶解于1,4-二恶烷/蒸馏水(25ml/46ml)后,加入氢氧化钠(0.5g,11.7mmol)和二碳酸二叔丁酯(3.0ml,12.9mmol),在氮大气、常温条件下,搅拌8小时。将蒸馏水(50ml)加入反应溶液后,利用乙酸乙酯(2×50ml)清洗。向水溶液层加入柠檬酸来进行酸性化,利用乙酸乙酯(3×50ml)提取后,用无水硫酸钠干燥。通过过滤及减压浓缩来得到化合物107(2.7g,68%)。
1h-nmr(400mhz,cdcl3)δ7.31(s,5h),5.15-5.01(m,2h),4.82-4.02(m,1h),3.68-3.43(m,2h),1.39(s,9h)。
化合物108的制备
将化合物107(2.7g,7.9mmol)溶解于甲醇(40ml)后,加入钯/木炭(10%)(pd/c,0.5g)。在常温、氢大气条件下,将反应溶液搅拌4小时。利用氟镁石过滤反应溶液,浓缩得到化合物108(1.2g,75%)。
1h-nmr(400mhz,d2o)δ3.70-3.65(m,1h),3.55-3.25(m,2h),1.28(s,9h)。
化合物109的制备
将化合物108(0.40g,1.96mmol)和马来酸酐(192mg,1.96mmol)溶解于乙酸(1.6ml)后,在常温条件下,搅拌3小时。减压浓缩反应溶液,将通过加入二氯甲烷(10ml)来生成的固体过滤后进行真空干燥。利用甲苯(15ml)稀释所干燥的该固体后,加入三乙胺(1.2ml,8.6mmol)和n,n-二甲基乙酰胺(0.75ml)来加热回流。反应16小时后,减压浓缩反应溶液,经高效液相色谱纯化后,通过冷冻干燥得到化合物109(287mg,52%)。
1h-nmr(400mhz,cdcl3)δ6.66(s,2h),5.17(br,1h),4.61(br,1h),3.68(br,1h),1.35(s,9h)。
化合物110的制备
将化合物109(0.15g,0.52mmol)溶解于n,n-二异丙基乙胺(3ml)后,加入n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(0.15g,0.79mmol)和n-羟基琥珀酰亚胺(0.09g,0.79mmol)。在常温条件下,将反应溶液搅拌12小时。将蒸馏水(30ml)加入反应溶液后,利用乙酸乙酯(30ml)提取。所提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物110(0.08g,40%)。
ei-msm/z:[m+na]+404.3。
实施例41.化合物112的制备
化合物111的制备
将化合物103(57mg,0.04mmol)和化合物110(0.016g,0.04mmol)溶解于n,n-二甲基甲酰胺(3ml)后,加入n,n-二异丙基乙胺(0.02ml,0.12mmol),在氮大气、常温条件下,搅拌6小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到化合物111(37mg,58%)。
ei-msm/z:[m+h]+1741.7,1/2[m+h]+871.7。
化合物112的制备
利用二氯甲烷(3ml)稀释化合物111(0.035g,0.02mmol)后,在0℃的温度下,加入三氟乙酸(0.3ml)搅拌3小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物112(6.5mg,20%)。
ei-msm/z:[m+h]+1641.9,1/2[m+h]+821.8。
实施例42.化合物115的制备
化合物114的制备
将2-[2-[2-(2-叠氮基甲氧基)乙氧基]乙氧基]乙酸(1.1g,4.71mmol)溶解于二氯甲烷(10ml)后,在0℃的温度、氮大气条件下,依次加入1-羟基苯并三唑(0.75g5.60mmol)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(1.15g,6.03mmol)后,搅拌30分钟。在氮大气条件下,加入溶解于二氯甲烷(10ml)的化合物113(1.5g,4.31mmol,化合物113利用wo2017/160569a1中描述的方法制备)和三乙胺(1.08ml,7.76mmol)溶液。将反应温度升温至常温并搅拌12小时后,利用二氯甲烷(100ml)稀释,用饱和碳酸氢钠水溶液(100ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物114(2.1g,87%)。
1h-nmr(400mhz,cdcl3)δ6.83(d,1h),4.15-4.11(m,1h),3.96(s,2h),3.72-3.62(m,14h),3.39-3.37(m,2h)1.81-1.60(m,4h),0.88(s,18h)0.42(s,12h)。
化合物115的制备
将化合物114(2.1g,3.73mmol)溶解于甲醇(30ml)后,在0℃的温度、氮大气条件下,加入浓盐酸(0.5ml)后,在常温条件下,搅拌2小时。利用三乙胺中和反应溶液后,浓缩,经柱层析纯化得到化合物115(1.2mg,98%)。
1h-nmr(400mhz,cdcl3)δ7.54(brs,1h),4.31-4.28(m,1h),4.02(s,2h),3.68-3.65(m,14h),3.43-3.40(m,2h),3.21(brs,2h),1.93-1.85(m,2h),1.64-1.57(m,2h)。
实施例43.化合物118的制备
化合物117的制备
将化合物116(1.32g,4.73mmol,化合物116利用pct/us2016/063564中描述的方法制备)溶解于二氯甲烷(20ml)后,在0℃的温度、氮大气条件下,加入1-羟基苯并三唑(0.86g,5.59mmol)和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(1.15g,6.02mmol)。在0℃的温度、氮大气条件下,加入溶解于二氯甲烷(5ml)的化合物113(1.5g,4.31mmol)和三乙胺(1.08ml,7.74mmol)溶液。将反应温度升温至常温搅拌12小时后,利用二氯甲烷(100ml)稀释,利用饱和碳酸氢钠水溶液(100ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物117(2.05g,79%)。
1h-nmr(400mhz,cdcl3)δ7.60(s,1h),6.92(d,j=9.2hz,1h),4.15-4.10(m,1h),4.05-4.03(m,2h),3.97(s,1h),3.73-3.66(m,10h),1.84-1.72(m,4h),1.48(s,9h),0.89(m,18h),0.05(s,12h)。
化合物118的制备
将化合物117(2.05g,3.37mmol)溶解于甲醇(10ml)后,在0℃的温度、氮大气条件下,加入樟脑磺酸(158mg,0.68mmol)后,在0℃的温度下,搅拌4小时。利用三乙胺(1ml)中和反应溶液后,浓缩,经柱层析纯化得到化合物118(1.28g,99%)。
1h-nmr(400mhz,cdcl3)δ7.76(d,j=7.2hz,1h),7.64(s,1h),4.32-4.29(m,1h),4.05-4.03(m,4h),3.75-3.69(m,10h),3.48(brs,2h),1.94-1.85(m,2h),1.68-1.61(m,4h),1.48(s,9h)。
实施例44.化合物124的制备
化合物119的制备
将3,5-吡唑二甲酸水合物(5g,28.71mmol)溶解于甲醇(50ml)后,在0℃的温度、氮大气条件下,加入亚硫酰氯(6.28ml,86.15mmol)后,加热至80℃。将反应溶液搅拌4小时后,通过浓缩得到化合物119(7.1g,99%)。
1h-nmr(400mhz,cdcl3)δ7.34(s,1h),3.96(s,6h)。
化合物120的制备
将化合物119(3.8g,20.63mmol)溶解于四氢呋喃(200ml)后,在0℃的温度、氮大气条件下,加入氢化铝锂(1m的四氢呋喃溶液,41.2ml,41.26mmol)后,加热回流12小时。将反应混合物降温至0℃,逐渐加入蒸馏水(50ml)后浓缩,利用甲醇(200ml)稀释后,再次加热至80℃。过滤热反应物,浓缩滤液。用乙醇(10ml)稀释滤液后,添加盐酸(4n的1,4-二恶烷溶液,82.6ml,22.7mmol),并搅拌20分钟。将通过向反应溶液中加入二乙醚(200ml)来生成的固体过滤并干燥,从而得到化合物120(2.6g,78%)。
1h-nmr(400mhz,dmso-d6)δ6.32(s,1h),4.52(s,4h)。
化合物121的制备
将化合物120(2.59g,15.73mmol)溶解于n,n-二甲基甲酰胺(75ml)后,在0℃的温度、氮大气条件下,加入咪唑(5.35g,78.68mmol)和叔丁基二甲基氯硅烷(5.69g,37.8mmol)。将反应溶液搅拌4小时后,用乙酸乙酯(100ml)稀释后,依次用饱和氯化铵水溶液(100ml)、盐水(100ml)洗涤,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物121(4.56g,81%)。
1h-nmr(400mhz,cdcl3)δ6.09(s,1h),4.74(s,4h),0.88(s,18h),0.09(s,12h)。
化合物122的制备
将化合物121(1.6g,4.48mmol)溶解于n,n-二甲基甲酰胺(25ml)后,在0℃的温度、氮大气条件下,加入碳酸铯(3.2g,9.8mmol)和三甘醇二甲苯磺酸酯(4.05g,8.97mmol)后,加热至50℃。将反应溶液搅拌4小时后,用乙酸乙酯(100ml)稀释,然后用盐水(100ml)洗涤,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物122(1.69g,60%)。
1h-nmr(400mhz,cdcl3)δ7.78(d,j=8hz,2h),7.33(d,j=8hz,2h),6.10(s,1h),4.67(d,j=5.2hz,4h),4.25-4.22(m,2h),4.13-4.11(m,2h),3.79-3.76(m,2h),3.62-3.60(m,2h),3.49-3.48(m,2h),3.46-3.45(m,2h),0.89(d,j=18hz,18h),0.06(d,j=5.6hz,12h)。
化合物123的制备
将化合物122(1.69g,2.62mmol)溶解于乙腈(25ml)后,在0℃的温度、氮大气条件下,加入n-羟基氨基甲酸叔丁酯(1.35g,5.51mmol)和1,8-二氮杂双环[5.4.0]-7-十一碳烯(0.8ml,5.38mmol)后,加热至50℃。将反应溶液搅拌12小时后,用乙酸乙酯(100ml)稀释,然后用盐水(100ml)洗涤,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物123(1.5g,60%)。
1h-nmr(400mhz,cdcl3)δ6.14(s,1h),4.70(d,j=8hz,4h),4.29-4.26(m,2h),3.83-3.74(m,4h),3.63-3.62(m,2h),3.55-3.53(m,2h),1.49(s,9h),0.92(d,j=18hz,18h),0.09(d,j=0.8hz,12h)。
化合物124的制备
将化合物123(1.5g,2.13mmol)溶解于四氢呋喃(20ml)后,在0℃的温度、氮大气条件下,加入四丁基氟化铵(1m的四氢呋喃溶液,8.18ml,8.18mmol)。将反应溶液搅拌12小时后,用乙酸乙酯(50ml)稀释,用饱和氯化铵水溶液(50ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物124(660mg,66%)。
1h-nmr(400mhz,cdcl3)δ6.22(s,1h),4.65(d,j=4.8hz,2h),4.57(d,j=6hz,2h),4.34-4.31(m,2h),3.38-3.38(m,2h),3.66-3.54(m,8h),1.52(s,9h)。
实施例45.化合物131的制备
化合物126的制备
将化合物125(1.12g,5.67mmol,化合物125利用wo2016/148674a1中描述的方法制备)溶解于二氯甲烷(30ml)后,在0℃的温度、氮大气条件下,加入吡啶(0.67ml,8.51mmol)和氯甲酸丙烯酯(0.66ml,6.24mmol)后,搅拌1小时。浓缩反应溶液后,经柱层析纯化得到化合物126(1.17g,73%)。
1h-nmr(400mhz,cdcl3)δ10.52(s,1h),8.06(s,1h),7.44(s,1h),6.07(s,1h),6.02-5.92(m,1h),5.36(d,j=17.2hz,1h),5.24(d,j=10.4hz,1h),4.66(d,j=5.2,2h),3.89(s,6h)。
化合物127的制备
将化合物115(920mg,2.75mmol)、化合物126(1.7g6.05mmol)和三苯基膦(2.52g,9.35mmol)溶解于干燥的四氢呋喃(20ml)后,在0℃的温度、氮大气条件下,加入偶氮二甲酸二异丙酯(1.66ml,8.52mmol)后,搅拌2小时。浓缩,经柱层析纯化得到化合物127(1.54g,65%)。
1h-nmr(400mhz,cdcl3)δ10.54(s,2h),8.07(s,2h),7.41(s,2h),6.00-5.93(m,2h),5.36(d,j=17.2hz,2h),5.25(d,j=10.0hz,2h),4.64-4.63(m,4h),4.44(bs,1h),4.23-4.20(m,4h),3.99(s,2h),3.89(s,3h),3.83(s,3h),3.66-3.60(m,11h),3.35-3.34(m,2h),2.25-2.13(m,4h)。
化合物128的制备
将化合物127(1.54g,1.78mmol)溶解于甲醇/四氢呋喃/蒸馏水(15ml/15ml/15ml)后,加入氢氧化钠(0.28g,7.15mmol)后,在40℃的温度下,搅拌5小时。用乙酸乙酯(100ml)稀释反应溶液后,用蒸馏水(100ml)提取。利用1n的盐酸水溶液酸性化合并的水溶液层后,用乙酸乙酯(100ml)提取后,用无水硫酸钠干燥。过滤浓缩得到化合物128(1.48g)。
1h-nmr(400mhz,dmso-d6)δ10.84(s,2h),7.94(s,2h),7.40(s,2h),6.00-5.95(m,2h),5.34(d,j=17.2hz,2h),5.24(d,j=10.0hz,2h),4.64-4.63(m,4h),4.18(brs,1h),4.04-4.01(m,4h),3.88(s,2h),3.74(s,6h),3.55-3.51(m,12h),2.05-1.98(m,4h)。
化合物129的制备
将化合物128(1.63g,1.95mmol)溶解于n,n-二甲基甲酰胺(5ml)后,在0℃的温度、氮大气条件下,加入n,n,n',n'-四甲基-o-(1h-苯并三唑-1-基)六氟磷酸脲(2.22g,5.87mmol)后,搅拌30分钟。在氮大气下,加入将溶于n,n-二甲基甲酰胺(5ml)的化合物4(0.64g,4.3mmol)和n,n-二异丙基乙胺(1.7ml,9.78mmol)溶液。将反应温度升温至常温并搅拌12小时后,用乙酸乙酯(100ml)稀释,用饱和碳酸氢钠水溶液(200ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物129(1.2g)。
1h-nmr(400mhz,cdcl3)δ8.65(brs,2h),7.34(brs,2h),7.21(d,j=8.8hz,1h),6.75(s,2h),6.00-5.90(m,2h),5.35(d,j=16.8hz,2h),5.23(d,j=10.4hz,2h),5.00-4.92(m,4h),4.68-4.57(m,6h),4.48-4.40(m,1h),4.20-4.08(m,8h),3.97(s,2h),3.79(s,6h),3.67-3.63(m,14h),3.39-3.37(m,2h),2.80-2.72(m,2h),2.48-2.44(m,2h),2.22-2.17(m,2h),2.10-2.04(m,2h)。
化合物130的制备
将化合物129(1.2g,1.17mmol)溶解于二氯甲烷(10ml)后,在0℃的温度、氮大气条件下,加入咪唑(0.4g,5.86mmol)和叔丁基二甲基氯硅烷(0.53g,3.5mmol)。将反应溶液搅拌12小时后,将盐水(50ml)加入反应溶液,用二氯甲烷(2×100ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物130(0.98g,3个步骤40%)。
1h-nmr(400mhz,cdcl3)δ8.81(brs,2h),7.77(s,2h),7.12(d,j=8hz,1h),6.80(s,2h),5.99-5.89(m,2h),5.34(d,j=17.2hz,2h)5.23(d,j=10.4hz,2h),4.98-4.91(m,4h),4.65-4.56(m,6h),4.54-4.44(m,1h),4.19-4.14(m,8h)4.01(s,2h),3.80(s,6h),3.66-3.61(m,14h),3.39-3.36(m,2h),2.69(s,4h),2.28-2.19(m,2h),2.15-2..05(m,2h),0.87(s,18h),0.03(s,12h)。
化合物131的制备
将化合物130(0.98g,0.78mmol)溶解于二氯甲烷(5ml)后,加入吡咯啶(0.16ml,1.95mmol)和四(三苯基膦)钯(0)(18mg,0.015mmol),在常温、氮大气条件下,搅拌6小时。将蒸馏水(50ml)加入反应溶液,用二氯甲烷(50ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物131(0.59g,70%)。
1h-nmr(400mhz,cdcl3)δ7.12.(d,j=9.2hz,1h),6.73(s,2h),6.26(s,2h),4.96-4.90(m,4h),4.52(bs,1h),4.38-4.35(m,4h),4.21-4.17(m,2h),4.11-4.03(m,6h),4.00(s,2h),3.75(s,6h),3.66-3.61m,12h),3.37-3.34(m,2h),2.7-2.68(m,4h)2.21-2.18(m,2h),2.12-2.05(m,2h),0.87(s,18h),0.02(s,12h)。
实施例46.化合物135的制备
化合物132的制备
将化合物131(590mg,0.54mmol)溶解于干燥的四氢呋喃(5ml)后,在-10℃的温度下,加入三光气(116mg,0.39mmol)和三乙胺(0.2ml,1.47mmol),在氮大气条件下,搅拌1小时。将化合物20(707mg,1.30mmol)溶解于干燥的四氢呋喃(5ml),加入三乙胺(0.2ml,01.47mmol)后,将该溶液缓慢加入反应溶液。1小时后,加热回流反应溶液并搅拌12小时。利用乙酸乙酯(30ml)稀释反应溶液后,用盐水(20ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物132(1.0g,83%)。
ei-msm/z:[m+h]+2219.10,1/2[m+h]+1110.30。
化合物133的制备
将化合物132(1g,0.45mmol)溶解于四氢呋喃/蒸馏水(5ml/5ml),加入乙酸(15ml)后,在常温、氮大气条件下,搅拌16小时。利用乙酸乙酯(50ml)稀释反应溶液后,用蒸馏水(50ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物133(720mg,80%)。
ei-msm/z:[m+h]+1990.95,1/2[m+h]+996.06。
化合物134的制备
将化合物133(370mg,0.18mmol)溶解于二氯甲烷(10ml)后,加入戴斯-马丁试剂(dess-martinperiodinane,181mg,0.42mmol),在常温、氮大气条件下,搅拌16小时。利用二氯甲烷(20ml)稀释反应溶液后,用饱和碳酸氢钠水溶液(20ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物134(350mg,90%)。
ei-msm/z:[m+h]+1986.61,1/2[m+h]+994.11。
化合物135的制备
将化合物134(350mg,0.17mmol)溶解于甲醇/四氢呋喃(7.5ml/7.5ml)后,在-40℃的温度下,缓慢加入溶解于蒸馏水(7.5ml)的氢氧化锂(66mg,1.58mmol)。将反应温度缓慢升温至0℃,搅拌2小时。用乙酸中和反应溶液后,减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物135(150mg,50%)。
ei-msm/z:[m+h]+1706.20,1/2[m+h]+854.00。
实施例47.化合物137的制备
化合物136的制备
化合物136利用化合物126和化合物118并通过类似于化合物134的合成的方法制备。ei-msm/z:[m+h]+2032.98,1/2[m+h]+1017.03。
化合物137的制备
将化合物136(205mg,0.10mmol)溶解于甲醇/四氢呋喃(4ml/6ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(38mg,0.91mmol)溶解于蒸馏水(4ml)的溶液。将反应温度逐渐升温至0℃并搅拌4小时。用乙酸中和反应溶液后,减压浓缩反应溶液并冷冻干燥。利用二氯甲烷(5ml)稀释所得到的固体后,在0℃的温度下,加入三氟乙酸(1.5ml)并搅拌4小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物137(29mg,17%)。
ei-msm/z:[m+h]+1653.01,1/2[m+h]+826.89。
实施例48.化合物138的制备
化合物138利用化合物126和化合物124并通过类似于化合物137的合成的方法制备。ei-msm/z:[m+h]+1647.60,1/2[m+h]+824.31。
实施例49.化合物142的制备
化合物139的制备
将二甲基亚砜(3.53ml,3.88mmol)溶解于二氯甲烷(30ml)后,在-78℃的温度、氮大气条件下,加入草酰氯(2.0m二氯甲烷溶液,13ml,23.9mmol)。将化合物6(6.8g,9.94mmol)溶解于二氯甲烷(20ml)后,在-78℃的温度、氮大气条件下,缓慢加入。将反应溶液搅拌10分钟后,将温度升温至0℃,缓慢加入三乙胺(13.85ml,4.41mmol)后,在常温条件下,搅拌2小时。将饱和氯化铵水溶液(200ml)加入反应溶液,利用二氯甲烷(3×100ml)提取。利用盐水(2×100ml)洗涤合并的有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物139(5.76g,85%)。
1h-nmr(400mhz,cdcl3)δ9.80(s,2h),7.72(s,2h),6.85(s,2h),5.07(s,2h),4.93(s,2h),4.22-4.10(m,6h),4.00(s,6h),3.94-3.80(m,4h),3.15-2.70(m,4h),2.30-2.10(m,2h),2.12-1.90(m,4h),1.75-1.68(m,6h).ei-msm/z:[m+h]+681.6。
化合物140的制备
将化合物139(1.84g,2.71mmol)溶解于苯和n,n-二甲基甲酰胺(v/v=10:1,30ml)后,在常温、氮大气条件下,依次加入乙二醇(1.5ml,27.11mmol)和樟脑磺酸(251mg,0.81mmol)。将反应溶液搅拌5分钟后,利用迪安-斯塔克(dean-stark)装置进行加热回流并搅拌2小时。浓缩反应溶液,利用乙酸乙酯(100ml)稀释后,将饱和碳酸氢钠水溶液(100ml)加入反应溶液,用乙酸乙酯(3×100ml)提取。利用盐水(2×100ml)洗涤合并的有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物140(1.53g,72%)。
1h-nmr(400mhz,cdcl3)δ7.70(s,2h),6.78(s,2h),5.07(s,2h),4.81(s,2h),4.69(s,2h),4.20-4.02(m,6h),4.00-3.90(m,8h),3.87-3.80(m,2h),3.78-3.70(m,4h),3.60-3.68(m,4h),2.72-2.60(m,4h),2.12-1.92(m,4h),1.65-1.60(m,2h).ei-msm/z:[m+h]+769.8。
化合物141的制备
将化合物140(1.11g,1.45mmol)溶解于乙醇(22ml)后,加入锌粉末(zincdust,2.84g,43.39mmol)和甲酸(5%的乙醇溶液,1.96ml)。在常温条件下,将反应溶液搅拌3小时后,利用氟镁石过滤,加入乙酸乙酯(300ml)。依次用蒸馏水(2×100ml)、饱和碳酸氢钠水溶液(2×200ml)和盐水(200ml)洗涤有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物141(800mg,78%)。
1h-nmr(400mhz,cdcl3)δ6.78(s,2h),6.23(s,2h),5.12(s,2h),4.97(s,2h),4.91(s,2h),4.78(brs,2h),4.54(brs,2h),4.33-4.21(m,2h),4.10-3.91(m,12h),3.90-3.82(m,4h),3.78(s,6h),2.72-2.58(m,4h),1.98-1.84(m,4h),1.74-1.58(m,2h),0.87(s,18h),0.02(s,12h)。ei-msm/z:[m+h]+709.8。
化合物142的制备
将化合物141(640mg,0.90mmol)溶解于二氯甲烷(45ml)后,在-78℃的温度、氮大气条件下,加入吡啶(0.15ml,1.80mmol)和氯甲酸丙烯酯(86μl,0.81mmol)。将反应溶液搅拌1小时后,将反应温度升温至常温,浓缩后,经柱层析纯化得到化合物142(320mg,43%)。
1h-nmr(400mhz,cdcl3)δ8.75(br,1h),7.83(s,1h),6.83(s,1h),6.77(s,1h),6.23(s,1h),5.97-5.91(m,1h),5.34,(d,j=17.2hz,1h),5.23(d,j=10.0hz,1h),5.08(brs,1h),5.02-4.88(m,6h),4.80(brs,1h),4.68-4.56(m,2h),4.44(brs,2h),4.30-4.18(m,2h),4.16-4.06(m,3h),3.92-3.84(m,3h),3.83(s,3h),3.78(s,3h),2.74-2.56(m,4h),1.99-1.86(m,4h),1.72-1.60(m,2h)。
实施例50.化合物146的制备
化合物143的制备
将化合物142(260mg,0.35mmol)溶解于四氢呋喃(4ml)后,在-10℃的温度下,加入三光气(40mg,0.13mmol)和三乙胺(0.078ml,0.56mmol),在氮大气条件下,搅拌1小时。将化合物20(208mg,0.39mmol)和三乙胺(0.087ml,0.62mmol)溶解于干燥的四氢呋喃(3ml),将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌3小时。浓缩反应溶液,用二氯甲烷(50ml)稀释后,用盐水(2×20ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物143(340mg,71%)。
1h-nmr(400mhz,cdcl3)δ8.78(brs,1h),7.95(d,j=12.2hz,1h),7.83(brs,1h),7.52-7.41(m,2h),7.27(s,1h),7.04(d,j=8.4hz,1h),6.83(s,1h),6.00-5.88(m,1h),5.42-5.23,(m,10h),5.20-5.08(m,4h),5.06-4.82(m,8h),4.67(s,1h),4.28-4.18(m,6h),4.16-4.06(m,8h),4.05-3.86(m,6h),3.86(s,3h),3.76(s,3h),3.52-3.62(m,3h),3.41(s,3h),2.78-2.58(m,2h),2.12-2.06(m,2h),2.05(s,9h),1.86-2.01(m,4h),1.72-1.60(m,2h),1.27(t,j=7.2hz,2h).ei-msm/z:[m+h]+1361.5,1/2[m+h]+681.6。
化合物144的制备
将化合物143(330mg,0.24mmol)溶解于二氯甲烷(5ml)后,加入吡咯啶(0.026ml,0.365mmol)和四(三苯基膦)钯(0)(14mg,0.012mmol),在常温、氮大气条件下,搅拌5小时。减压浓缩反应溶液后,经柱层析纯化得到化合物144(290mg,90%)。
1h-nmr(400mhz,cdcl3)δ8.82(brs,1h),8.05(s,1h),7.52-7.42(m,2h),7.04(d,j=8.2hz,1h),6.82(s,1h),6.78(s,1h),6.24(s,1h),5.44-5.26,(m,4h),5.16-5.04(m,4h),5.02-4.86(m,5h),4.52-4.38(m,2h),4.30-4.10(m,6h),4.16-4.07(m,5h),4.04-3.92(m,6h),3.91-3.84(m,6h),3.83(s,3h),3.78(s,3h),3.72(s,3h),3.60-3.52(m,3h),3.41(s,3h),2.71-2.58(m,4h),2.05(s,9h),1.97-1.85(m,4h),1.72-1.58(m,4h),1.25(t,j=7.2hz,2h).ei-msm/z:[m+h]+1277.2,1/2[m+h]+639.4。
化合物145的制备
将化合物144(340mg,0.29mmol)溶解于干燥的四氢呋喃(3ml)后,在-10℃的温度下,加入三光气(25mg,0.09mmol)和三乙胺(0.060ml,0.43mmol),在氮大气条件下,搅拌1小时。将化合物32(229mg,0.31mmol)溶解于干燥的四氢呋喃(3ml),加入三乙胺(0.060ml,0.43mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(100ml)稀释后,用盐水(2×50ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物145(250mg,43%)。
ei-msm/z:[m+na]+2056.4,1/2[m+h]+967.7。
化合物146的制备
将化合物145(230mg,0.113mmol)溶解于甲醇/四氢呋喃(3ml/3ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(48mg,1.13mmol)溶解于蒸馏水(6ml)的溶液。将反应温度缓慢升温至0℃并搅拌2小时。用乙酸中和反应溶液后,减压浓缩并干燥反应溶液。利用二氯甲烷(10ml)稀释所得到的固体后,在0℃的温度下,加入三氟乙酸(2ml)并搅拌2小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色的固体化合物146(15.6mg)。
ei-msm/z:[m+h]+1653.7,1/2[m+h]+827.6。
实施例51.化合物148的制备
化合物147的制备
将化合物144(266mg,0.21mmol)溶解于干燥的四氢呋喃(3ml)后,在-10℃的温度下,加入三光气(16mg,0.06mmol)和三乙胺(0.044ml,0.31mmol),在氮大气条件下,搅拌1小时。将化合物55(147mg,0.23mmol)溶解于干燥的四氢呋喃(3ml),加入三乙胺(0.044ml,0.31mmol)后,将该溶液缓慢加入反应溶液。30分钟后,加热回流反应溶液并搅拌4小时。浓缩反应溶液,利用二氯甲烷(100ml)稀释后,用盐水(2×50ml)洗涤,用无水硫酸钠干燥。过滤及减压浓缩后,经柱层析纯化得到化合物147(170mg,42%)。
ei-msm/z:[m+na]+1944.6,1/2[m+h]+972.8。
化合物148的制备
将化合物147(120mg,0.087mmol)溶解于甲醇/四氢呋喃(3ml/3ml)后,在-40℃的温度下,缓慢加入将氢氧化锂(37mg,0.87mmol)溶解于蒸馏水(6ml)的溶液。将反应温度缓慢升温至0℃并搅拌2小时。用乙酸中和反应溶液后,减压浓缩并干燥反应溶液。利用二氯甲烷(8ml)稀释所得到的固体后,在0℃的温度下,加入三氟乙酸(2ml)并搅拌2小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物148(31mg)。
ei-msm/z:[m+h]+1663.4,1/2[m+h]+832.7。
实施例52.化合物155的制备
化合物149的制备
将化合物4(13.8g,92.5mmol)溶解于二氯甲烷(400ml)后,在0℃的温度、氮大气条件下,加入溶解有咪唑(18.8g,277.5mmol)和二氯甲烷(100ml)的叔丁基二甲基氯硅烷(15.3g,101.7mmol)。在常温条件下,将反应溶液搅拌2小时后,将盐水(30ml)加入反应溶液,用二氯甲烷(2×300ml)提取后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物149(17g,80%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ4.91,(d,j=14.4hz,2h),3.66-3.47(m,4h),3.27-3.24(m,1h),2.47-2.42(m,1h),2.24-2.18(m,1h),0.91(s,9h),0.05(s,6h)。
化合物151的制备
将化合物150(17.3g,46.8mmol,化合物150利用acsmed.chem.lett.2016,7,983中描述的方法制备)溶解于n,n-二甲基甲酰胺(100ml)后,在0℃的温度、氮大气条件下,依次加入1-羟基苯并三唑(6.8g50.7mmol)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(10.4g,54.6mmol)后,搅拌30分钟。在氮大气下,加入溶解于二氯甲烷(50ml)的化合物149(8.8g,39.0mmol)和三乙胺(9.78ml,70.2mmol)溶液。将反应温度升温至常温,搅拌12小时后,利用二氯甲烷(100ml)稀释,用饱和碳酸氢钠水溶液水溶液(100ml)洗涤后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物151(19.9g,89%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.69(s,1h),6.72(s,1h),4.97(s,1h),4.82(s,1h),4.57-4.54(m,1h)3.89(s,4h),3.74-3.71(m,2h)3.30-3.27(m,1h),2.76-2.52(m,2h),1.31-1.23(m,3h),1.08(s,18h),0.89(s,9h),0.08(s,3h)。
化合物152的制备
将化合物151(29.5g,50.9mmol)溶解于乙醇(720ml)后,加入锌粉末(zincdust,66.6g,1019.1mmol)和甲酸(38ml,1019.1mmol)。在常温条件下,将反应溶液搅拌40分钟后,利用氟镁石过滤,加入乙酸乙酯(500ml)。依次用蒸馏水(500ml)、饱和碳酸氢钠水溶液(500ml)和盐水(500ml)洗涤有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物152(27.9g,99%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ6.71(s,1h),6.25(s,1h),4.96-4.89(m,2h),4.53(brs,1h),4.21-4.09(m,4h),3.74(brs,1h),3.71(s,3h),3.62(brs,1h),2.73-2.63(m,2h),1.29-1.21(m,3h),1.05(s,18h),0.87(s,9h),0.02(s,6h)。
化合物153的制备
将化合物152(27.9g,50.8mmol)溶解于二氯甲烷(300ml)后,在-78℃的温度、氮大气条件下,加入吡啶(9ml,111.8mmol)和氯甲酸丙烯酯(5.9ml,55.9mmol)。将反应溶液搅拌1小时后,将反应温度升温至常温,浓缩后,经柱层析纯化得到化合物153(31.8g,99%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ8.67(brs,1h),7.75(s,1h),6.78(s,1h),5.99-5.89(m,1h),5.33,(d,j=17.2hz,1h),5.21(d,j=10.4hz,1h),4.98-4.90(m,2h),4.66-4.57(m,3h),4.19-4.11(m,1h),4.01(brs,1h),3.86(brs,1h),3.76(s,3h),3.65(brs,1h),2.68(s,2h),1.33-1.24(m,3h),1.05(s,18),0.87(s,9h),0.03(s,6h)。
化合物154的制备
将化合物153(31.8g,50.2mmol)溶解于n,n-二甲基甲酰胺(300ml)和蒸馏水(6ml)后,在0℃的温度、氮大气条件下,加入乙酸钠(5g,60.2mmol),并在常温条件下,搅拌2小时。利用乙酸乙酯(300ml)稀释反应溶液后,用蒸馏水(300ml)洗涤,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物154(17.7g,74%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ8.75(brs,1h),7.75(s,1h),6.78(s,1h),6.14(s,1h),5.94-5.90(m,1h),5.32,(d,j=17.2hz,1h),5.21(d,j=10.4hz,1h),4.97-4.90(m,2h),4.65-4.56(m,3h),4.18-4.15(m,1h),4.01(brs,1h),3.85(s,4h),3.65(brs,1h),2.68(s,2h),0.87(s,9h),0.02(s,6h)。
化合物155的制备
将化合物154(18.6g,39.0mmol)溶解于丙酮(200ml)后,在氮大气下,依次加入1,5-二碘戊烷(11.6ml,156mmol)和碳酸钾(5.9g,42.9mmol)后,在60℃的温度下,搅拌12小时。浓缩反应溶液,经柱层析纯化得到化合物155(23g,87%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ8.88(brs,1h),7.83(s,1h),6.81(s,1h),5.98-5.90(m,1h),5.34,(d,j=17.2hz,1h),5.24(d,j=10.4hz,1h),4.98-4.90(m,2h),4.67-4.58(m,3h),4.21-4.12(m,1h),4.10-4.06(m,3h)3.82(s,4h),3.64(brs,1h),3.23-3.19(m,2h),2.69(s,2h),1.94-1.84(m,4h),1.62-1.55(m,2h),0.87(s,9h),0.03(s,6h)。
实施例53.化合物162的制备
化合物156的制备
将化合物151(9.3g,16.0mmol)溶解于n,n-二甲基甲酰胺(100ml)和蒸馏水(2ml)后,在0℃的温度、氮大气条件下,加入乙酸钠(1.6g,19.2mmol),在常温条件下,搅拌30分钟。利用乙酸乙酯(100ml)稀释反应溶液后,用蒸馏水(100ml)洗涤,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物156(5.4g,80%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.75(s,1h),6.76(s,1h),6.07(s,1h),4.98(s,1h),4.83(s,1h),4.58-4.54(m,1h),3.99(s,3h),3.89-3.87(m,1h),3.77-3.70(m,2h),3.33-3.29(m,1h),2.81-2.53(m,2h),0.89(s,9h),0.09(s,6h)。
化合物157的制备
将化合物156(3.0g,7.1mmol)溶解于n,n-二甲基甲酰胺(30ml)后,在0℃的温度、氮大气条件下,加入碳酸钾(1.1g,7.8mmol)和苄基溴(0.9ml,7.8mmol)。将反应溶液搅拌3小时后,将饱和氯化铵水溶液(50ml)加入反应溶液,用乙酸乙酯(2×50ml)提取。依次用蒸馏水(2×100ml)、盐水(100ml)洗涤合并的有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物157(3.6g,97%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.77(d,j=4.8hz,1h),7.46-7.33(m,5h),6.79(d,j=18.8hz,1h),5.22(d,j=5.2hz,2h),5.09(d,j=7.6hz,1h),4.98(s,1h),4.83(s,1h),4.58(brs,1h),3.96(s,3h),3.87(brs,1h),3.77-3.69(m,2h),3.30-3.28(m,1h),2.81-2.53(m,2h),0.89(s,9h),0.09(s,6h)。
化合物158的制备
将化合物157(3.6mg,6.9mmol)溶解于四氢呋喃/蒸馏水(15ml/15ml),加入乙酸(30ml)后,在常温、氮大气条件下,搅拌16小时。减压浓缩反应溶液后,经柱层析纯化得到化合物158(2.8g,99%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ7.77(s,1h),7.47-7.35(m,5h),6.81(s,h),5.22(s,2h),5.02(s,1h),4.87(s,1h),4.60(brs,1h),3.99(s,3h),3.88(brs,1h),3.83-3.72(m,3h),3.54(brs,1h),2.88-2.82(m,1h),2.52-2.48(m,1h)。
化合物159的制备
将草酰氯(2.1ml,14.1mmol)溶解于二氯甲烷(20ml)后,在-78℃的温度、氮大气条件下,加入二甲基亚砜(1.5ml,21.1mmol)。1小时后,缓慢加入将化合物158(2.7g,6.9mmol)溶解于二氯甲烷(50ml)的溶液。将反应溶液搅拌2小时后,将三乙胺(3.4ml,42.3mmol)稀释于二氯甲烷(30ml)并缓慢加入。经2小时将反应温度缓慢升温至0℃。利用二氯甲烷(100ml)稀释反应溶液,用饱和氯化铵水溶液(200ml)和盐水(200ml)洗涤有机层后,用无水硫酸钠干燥。过滤浓缩,经柱层析纯化得到化合物159(2.7g,96%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ9.79(s,1h),7.79(s,1h),7.46-7.26(m,5h),6.87(s,1h),5.22(s,2h),5.06-4.96(m,1h),4.93-4.90(m,1h),4.78(brs,1h),4.62-4.56(m,1h),3.99(s,3h),3.93(s,1h),3.85(s,1h),2.91-2.62(m,2h)。
化合物160的制备
将化合物159(2.7g,6.8mmol)溶解于四氢呋喃/蒸馏水(60ml/40ml)后,加入连二亚硫酸钠(na2s2o4,11.2g,64.4mmol)。在氮大气条件下,搅拌20小时。向反应溶液中加入甲醇(60ml)来稀释,加入6n的盐酸水溶液来进行酸性化(ph2)并搅拌1小时。减压浓缩反应溶液去除甲醇。加入6n的盐酸水溶液来进行酸性化(ph2),用乙酸乙酯(5×100ml)提取。用无水硫酸钠干燥合并的有机层。过滤浓缩,经柱层析纯化得到化合物160(1.8g,77%)。
ei-msm/z:[m+h]+349.3,[m+h2o]+367.3。
化合物161的制备
将化合物160(1.8g,5.3mmol)溶解于二氯甲烷/n,n-二甲基甲酰胺(20ml/8ml)后,在0℃的温度、氮大气条件下,加入三乙酰氧基硼氢化钠(1.2g,5.8mmol)后,搅拌2小时。将蒸馏水(40ml)加入反应溶液后,用二氯甲烷(2×50ml)提取。所提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物161(1.2g,64%)。
1h-nmr(400mhz,cdcl3)δ7.61(s,1h),7.41-7.30(m,5h),6.05(s,1h),5.12(s,2h),5.06(s,1h),5.02(s,1h),4.38(d,j=18hz,1h),4.27(d,j=16.4hz,1h),4.04-3.96(m,1h),3.86(s,3h),3.49(d,j=11hz,1h),3.29(dd,j=9.2hz,1h),2.91-2.85(m,1h),2.40(dd,j=10hz,1h)。
化合物162的制备
将化合物161(1.3g,3.7mmol)溶解于二氯甲烷(70ml)后,加入甲磺酸(25ml),在氮大气条件下,搅拌2小时。将蒸馏水(20ml)加入反应溶液后,加入碳酸钠来中和。向反应溶液中加入水(200ml)稀释,用二氯甲烷(3×50ml)提取。所提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物162(620mg,64%)。
1h-nmr(400mhz,cdcl3)δ7.60(s,1h),6.17(s,1h),5.88(brs,1h),5.09(s,1h),5.06(s,1h),4.41(d,j=16.4hz,1h),4.31(d,j=16.4hz,1h),4.08-3.99(m,1h),3.88(s,3h),3.54(d,j=12.4hz,1h),3.49(d,j=11hz,1h),3.34(dd,j=9.2hz,1h),2.95-2.89(m,1h),2.43(dd,j=6.4hz,1h)。
实施例54.化合物164的制备
化合物163的制备
将化合物162(374mg,1.4mmol)和化合物155(1.0g,1.5mmol)溶解于丙酮/n,n-二甲基甲酰胺(20ml/20ml)后,在氮大气条件下,加入碳酸钾(258mg,1.8mmol),在80℃的温度下,加热搅拌12小时。过滤反应溶液后减压浓缩,将蒸馏水(20ml)加入反应溶液后,用乙酸乙酯(3×30ml)提取。所提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物163(620mg,53%)。
1h-nmr(400mhz,cdcl3)(rotamers)δ8.86(brs,1h),7.85(s,1h),7.60(s,1h),6.82(s,1h),6.06(s,1h),6.01-5.91(m,1h),5.36(d,j=17.2hz,1h),5.25(d,j=10.4hz,1h),5.08(s,1h),5.05(s,1h),5.00(s,1h),4.92(brs,1h),4.63(d,j=4.8hz,2h),4.41(d,j=16.4hz,1h),4.30(d,j=16.4hz,1h),4.20(dj=14hz,1h),4.13-4.10(m,3h),4.05-3.98(m,3h),3.85(s,3h),3.82(s,3h),3.66(bs,1h),3.55(d,j=12.8hz,1h),3.32(dd,j=9.2hz,1h),2.91(dd,j=8.8hz,1h),2.70(brs,2h),2.43(dd,j=7.2hz,1h),1.97-1.91(m,4h),1.69-1.64(m,2h),0.89(s,9h),0.04(brs,6h)。
化合物164的制备
化合物164利用化合物163并通过类似于化合物28的合成的方法制备。ei-msm/z:[m+h]+1548,1/2[m+h]+775。
实施例55.化合物167的制备
化合物165的制备
将l-组氨酸(5.0g,32.22mmol)溶解于二氯甲烷(45ml)后,在常温条件下,加入二氯甲烷(3.9ml,32.22mmol)和三乙胺(9.0ml,64.44mmol),在氮大气条件下,将反应溶液加热回流4小时。加入三苯甲基氯(8.9g,32.22mmol)和三乙胺(4.5ml,32.22mmol),在氮大气条件下,搅拌2小时,将甲醇(50ml)加入反应溶液后减压浓缩,通过加入蒸馏水(50ml)和三乙胺来将ph调至8~8.5左右后,过滤未溶解的浆料,依次用氯仿(50ml)、二乙醚(50ml)和蒸馏水(50ml)清洗。干燥所形成的白色固体化合物得到化合物165(三乙胺盐,12.4g,95%)。
1h-nmr(400mhz,cd3od)δ7.45-7.32(m,10h),7.21-7.15(m,5h),3.75-3.77(m,1h),3.20(q,2h),3.00-2.97(m,1h),1.32(t,3h)。
化合物166的制备
将化合物165(1.0g,2.52mmol)和n-甲氧基羰基马来酰亚胺(429mg,2.77mmol)溶解于1,4-二恶烷/蒸馏水(5ml/2.5ml)后,加入碳酸钠(267mg,2.52mmol),在氮大气条件下,加热回流12小时。减压浓缩反应溶液,溶解于n,n-二甲基甲酰胺(3ml)后,将三乙胺(0.16ml,1.12mmol)加入反应溶液,在氮大气条件下,搅拌10小时。将蒸馏水(5ml)加入反应溶液后,通过加入0.5n的盐酸水溶液来进行酸性化(ph4),加入二氯甲烷(3×10ml)提取后,用无水硫酸钠干燥。经过滤及减压浓缩得到化合物166(504mg,32%)。
ei-msm/z:[m+h]+478.4,[m+na]+500.4。
化合物167的制备
将化合物166(252mg,0.53mmol)溶解于n,n-二异丙基乙胺(4ml)后,加入n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(132mg,0.69mmol)和n-羟基琥珀酰亚胺(85mg,0.74mmol)。在常温条件下,将反应溶液搅拌12小时。将蒸馏水(30ml)加入反应溶液后,用乙酸乙酯(2×30ml)提取。提取的有机层用无水硫酸钠干燥,过滤及减压浓缩后,经柱层析纯化得到化合物167(274mg,90%)。
ei-msm/z:[m+h]+575.3。
实施例56.化合物169的制备
化合物168的制备
将化合物103(50mg,0.03mmol)和化合物167(27.4mg,0.05mmol)溶解于n,n-二甲基甲酰胺(1ml)后,加入n,n-二异丙基乙胺(0.01ml,0.05mmol),在氮大气、常温条件下,搅拌12小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到化合物168(11mg,18%)。
ei-msm/z:[m+h]+1934.8,1/2[m+h]+968.0。
化合物169的制备
利用二氯甲烷(0.75ml)稀释化合物168(11mg,6μmol)和苯甲醚(6μl,60μmol)后,在0℃的温度下,加入三氟乙酸(0.25ml)并搅拌3小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到白色固体化合物169(2mg,21%)。
ei-msm/z:[m+h]+1692.7,1/2[m+h]+846.9。
实施例57.化合物171的制备
化合物170的制备
将dbco-peg4-酸(50mg,91μmol)溶解于二氯甲烷(1ml)后,加入n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(19mg,99μmol)和n-羟基琥珀酰亚胺(11mg,99μmol),在氮大气、常温条件下,搅拌3小时。减压浓缩反应溶液后得到化合物170(59mg)。
ei-msm/z:[m+h]+650.7。
化合物171的制备
将化合物103(47mg,32μmol)和化合物170(24mg,38μmol)溶解于n,n-二甲基甲酰胺/二氯甲烷(1ml/0.25ml)后,加入n,n-二异丙基乙胺(51μl,38μmol),在氮大气、常温条件下,搅拌3小时。减压浓缩反应溶液后,经高效液相色谱纯化,冷冻干燥得到化合物171(8.1mg,14%)。
ei-msm/z:[m+h]+2010.1,1/2[m+h]+1005.6。
化合物172至178的制备
下面,参照参考文献制备了如表1所示的结构的吡咯并苯并二氮杂二聚物化合物172至化合物178。
[表1]
实施例58
抗体药物缀合物的制备
抗体药物缀合物通过如下两个步骤来制备,共同适用的lcb14-0511、lcb14-0512以及lcb14-0606利用韩国公开专利第10-2014-0035393中描述的方法制备而成。lcb14-0606、lcb14-0511以及lcb14-0512的结构式如下:
步骤1:异戊烯基抗体(prenylatedantibody)的制备
制备抗体的异戊烯化反应混合物并在30℃的温度下使其反应16小时。反应混合物由24μm的抗体、包含200nm的法尼基蛋白转移酶(calbiochem#344145)及0.144mm的lcb14-0511或lcb14-0512或lcb14-0606的缓冲溶液(50mm的tris-hcl(ph7.4)、5mm的mgcl2、10μm的zncl2、0.5mm的dtt)构成。反应结束后,异戊烯化抗体利用由pbs缓冲溶液均衡化的g25sepharose柱(aktapurifier,gehealthcare)来制盐。
步骤2:药物结合(drug-conjugation)方法
<肟键反应(conjugationbyoximebondformation)>
异戊烯化抗体与连接体-药物之间的肟键生成反应混合物是通过混合ph为4.5的100mm的na-乙酸缓冲溶液、10%的dmso、24μm的抗体和240μm的连接体-药物(inhouse,实施例8-10及比较例1的最终产物为表1的化合物)来制备,并在30℃的温度下轻轻搅拌。反应24小时后,经快速蛋白液相色谱(fplc)(aktapurifier,gehealthcare)过程去除了所使用过的大量低分子化合物,收集蛋白质分馏并浓缩。
<点击结合反应(conjugationbyclickreaction)>
异戊烯化抗体与连接体-药物之间的点击(click)反应混合物是通过混合10%的dmso、24μm的抗体和240μm的连接体-药物(inhouse,实施例20、21、22、26、27、28、46的最终产物为表1的化合物)、1mm的五水合硫酸铜(ii)、2mm的(bimc4a)3(sigma-aldrich696854)、10mm的抗坏血酸钠(sodiumascorbate)以及10mm的氨基胍盐酸盐来制备,在25℃的温度下使其反应3小时后,处理2.0mm的edta并使其反应30分钟。反应结束后,经快速蛋白液相色谱(fplc)(aktapurifier,gehealthcare)过程去除了所使用过的大量低分子化合物,收集蛋白质分馏并浓缩。
[表2]
抗体药物缀合物制备目录
实验例1.生物体外(invitro)细胞毒性评价
测定了对于在下述表2中记载的药物及抗体药物缀合物的癌细胞株的细胞增殖抑制活性。作为癌细胞株使用了市售的人类乳腺癌细胞株mcf-7(her2阴性或正常)及sk-br3(her2阳性)、jimt-1(her2阳性)。作为药物,将mmaf-ome作为抗体药物缀合物,使用了表1的抗体药物缀合物。在96-孔板中,将各癌细胞株以每孔(well)接种5000~13000个72小时处理组、每孔接种1500~3000个168小时处理组来培养24小时后,以0.0051~33.33nm或0.0015~10.0nm(连续稀释3倍)的浓度处理抗体和抗体药物缀合物,以0.023~50nm(连续稀释3倍)的浓度处理药物。通过使用磺酰罗丹明b(sulforhodamineb,srb)染料定量经72/168小时之后存活的细胞数。
[表3]
抗体药物缀合物试样的细胞毒性比较
a.培养时间(incubationtime)/b.nt:没有测试(nottested)/c.培养144小时(144hincubation)
在sk-br3和jimt-1乳腺癌细胞株中,相对于抗体-药物缀合物22、23、24试样,抗体药物缀合物中导入有吡咯并苯并二氮杂的两个n10位置均由氨基甲酸酯结构组成的前体连接体-药物28、29、30的抗体药物缀合物1、2、3的细胞毒性非常优秀。
在以本发明的前体形态给药的情况下,相对于现有吡咯并苯并二氮杂药物,在以下方面有利,即,由于在血液中暴露时需要通过额外的反应转换为有效物质,因此可预先防止在意外的连接体分解时可能会发生的副作用的可能性,对于正常细胞的毒性降低,药物更加稳定。
并且,当制备抗体-药物缀合物时,在以现有方法制备的抗体-药物缀合物的情况下,存在杂质的含量高、暴露的亚胺基受亲核试剂攻击而生成不需要的结构的药物的隐患,相反,以本发明的方法制备的抗体-药物缀合物,由于吡咯并苯并二氮杂二聚物的亚胺基是前药的形式,因此具有免受亲核试剂攻击的优点,并且其纯度高,因此具有容易分离的优点,相对于现有吡咯并苯并二氮杂或吡咯并苯并二氮杂二聚物,物性进一步提高。
产业上的可利用性
本发明的吡咯并苯并二氮杂二聚物前体、吡咯并苯并二氮杂二聚物前体-连接体或吡咯并苯并二氮杂二聚物前体-连接体-配体缀合物可用于癌等增殖性疾病的靶向化、特异性治疗。
- 还没有人留言评论。精彩留言会获得点赞!
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗慢性阻塞性肺病药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗红细胞低下性贫血药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗病毒药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗菌药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗急性痛风药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗骨质疏松药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗鼻炎药物中的应用
- 一种(3aR,9bS)-叔丁基-3,3a,5,9b-四氢-1氢-吡咯[3,4-c]异喹啉-4(2氢)-羧酸酯的制 ...的制作方法
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在预防或治疗胰腺纤维化药物中的应用
- Virosaine A的四氢吡咯基和吗啉基衍生物的组合物在抗缺氧药物中的应用
- 一种具有抗氧化活性的芳基苯并呋喃类化合物及其制备方法和应用与流程
- 一种含苯并双环酮与乙氧磺隆的除草组合物及其应用的制造方法与工艺
- 一种6‑氯呋喃[3,2‑B]吡啶的合成方法与流程
- 一类苯并吡喃类化合物及其应用的制造方法与工艺
- 一种以二苯并环庚烯为核心的化合物及其在有机电致发光器件上的应用的制造方法与工艺
- 一种含苯并双环酮与乙草胺的除草组合物及其应用的制造方法与工艺
- 一种含苯并双环酮与噁草酮的除草组合物及其应用的制造方法与工艺
- 分解二噁英前体的催化剂筛选系统及其使用方法与流程
- 一种无卤热固性树脂组合物以及含有它的预浸料、层压板和印制电路板的制造方法与工艺
- 一种以二联二苯并五元杂环为骨架的有机化合物及其在OLED上的应用的制造方法与工艺