κ阿片受体拮抗剂以及与其相关的产品和方法与流程
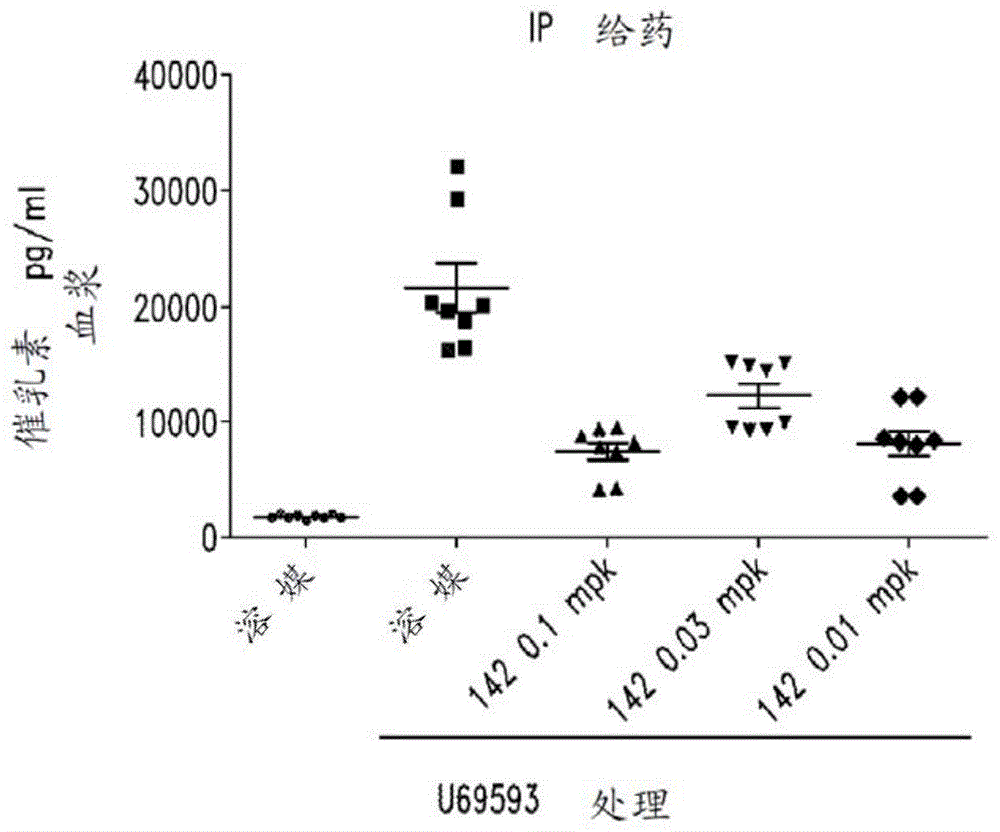
发明领域本发明涉及κ(kappa)-阿片受体(kor)拮抗剂和含有其的产品,以及其应用和制备方法。背景oprk1基因编码κ-阿片受体(kor),并且κ-阿片受体是阿片受体家族成员,其结合作为主要的内源性配体的阿片样肽强啡肽。kor在脑、脊髓和外周组织具有广泛而清晰的分布,并且特别分布于与奖励、认知功能和应激反应相关的脑部区域。证据表明在疼痛和应激条件下,强啡肽类升高,并且kor破坏产生抗应激作用。所述发现引起kor拮抗剂的研发,用于治疗抑郁、焦虑、成瘾性病患,以及与应激相关的其他精神病症。kor拮抗剂的研发总结于urbano等人题为“antagonistsofthekappaopioidreceptors”(κ阿片受体拮抗剂),bioorganic&medicinalchemistryletters,24:2021-2032,2014中。典型kor拮抗剂(即衍生自吗啡烷的配体nor-bni和gnti,以及非吗啡烷jdtic)的药理学研究已证实kor/强啡肽体系的治疗潜能。然而,所述典型kor拮抗剂显示数小时至数天的延迟显效,随后在最小有效剂量拮抗作用持续数周。此外,所述化合物显示低的血脑屏障透过。因此,更近的研究集中于具有改善的药代动力学的短效kor拮抗剂的研发。已广泛研究kor拮抗剂,因为其阻断显著应激引起的精神适应;即伏核(nac)中强啡肽的表达升高。nac是中脑边缘系统的一个元件,其在精神病的刺激和病理学中起作用。应激,以及反复暴露于药物滥用引起与nac中camp反应元件结合蛋白转录因子creb相关的一系列复杂胞内事件。根据carlezon等人在“kappaopioidantagonistsforpsychiatricdisorders:frombenchtoclinicaltrials”(用于精神病的κ阿片类拮抗剂:从实验室到临床试验)(depressionandanxiety,33:895-906,2016)中的解释,creb-介导的强啡肽表达增加产生抑郁样体征,kor拮抗剂减轻所述体征。根据carlezon等人提出的模型,应激激活nac中的creb,这致使强啡肽表达增加。增加的强啡肽反过来促进kors活化。kors表达在细胞体和中央皮质边缘(mesocorticolimbic)多巴胺(da)神经元的末端上,并且kors的活化抑制da释放。用kor拮抗剂治疗阻断强啡肽的作用,恢复da功能,并由此在各种动物模型内提供抗抑郁和抗焦虑样作用。所述作用机制,以及迄今kor拮抗剂相当广泛的研发和测试包括最近的临床研究结果(例如cerc-501和alks-5461),提供有力证据,即kor拮抗剂对患例如diagnosticandstatisticalmanualofmentaldisorder(dsm,精神障碍诊断和统计手册)中定义的广泛病患包括心境障碍、焦虑症和物质使用障碍(substanceusedisorders)的人可具有治疗作用。将精神病分类的另一架构是researchdomaincriteria(研究领域标准,rdoc)项目,其目标是根据可见的行为尺度和神经生物学尺度,将所述病患分类。在该环境中,kor拮抗剂对至少两种类型的rdoc-定义的区域具有治疗作用;即与奖赏相关的和与应激的副作用相关的那些。在这些区域内,已公认将kor拮抗剂用于治疗快感缺乏(“正价体系”),以及用于阻断应激的副作用(“负价体系”)。根据在所述领域取得的进展,认为将kor拮抗剂用于治疗重性抑郁,以及与物质滥用或成瘾性相关的病患,特别是在快速显效治疗环境中,这避免与上文讨论的典型kor拮抗剂相关的缺点。其他研究已显示kor拮抗剂会是特别适合用于治疗应激介导的症状,以及用于治疗社交焦虑障碍和恐怖症。预防治疗也已表明预防应激带来的不良情况,并且在这点上,已将kor拮抗推荐在具有患所述疾病风险的个体中,作为ptsd的预防治疗。kor拮抗的其他治疗用途包括奖赏相关功能损害的治疗,因为所述损害经常发生于患心境和焦虑谱障碍的患者中,并且所述损害还可伴随其他类型的病症例如精神分裂症或情感性分裂症而发生。总之,kor拮抗为多种病症和病患的治疗提供重大前景。因此,一些化合物目前在研发中,其是高度选择性和有效的kor拮抗剂,用于治疗各种病患例如物质使用障碍、重性抑郁、快感缺乏和应激相关的症状。然而,尽管在本领域取得的进展,还需要新的和改善的kor拮抗剂和含有其的药品,以及与其应用和制备相关的方法。发明简述本发明涉及拮抗κ阿片受体(kor)的化合物,含有其的组合物,及其制备方法和治疗疾病或病患的用途,其中kor的拮抗是医学上指示的或有益的。在一实施方案中,提供具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐:其中x、y、r1、r2、r4、r5、r6、r7、r8和r11是下文定义的。在一实施方案中,提供药物组合物,其包含具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,以及药学上可接受的载体、稀释剂或赋形剂。在一实施方案中,提供具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐用于制备药物的用途。在一实施方案中,提供拮抗kor的方法,所述方法包含使受体与有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物接触。在一实施方案中,提供治疗神经精神病学病患或行为病患的方法,无论器质性的、应激诱导的或医源性的,所述病患特征为血清催乳素的升高,以及对kor拮抗剂施用响应伴随血清催乳素降低。所述方法包括以足以为个体提供有益作用的频率和持续期,向个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供在医学上指示的kor拮抗的个体中治疗不良状况(malcondition)的方法。所述方法包括以足以为个体提供有益作用的频率和持续期,向个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗成瘾症(addictivedisorder)包括与物质滥用或成瘾相关病症的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗中枢神经系统(cns)-相关病症的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗焦虑症的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,焦虑症是社交焦虑症。在一实施方案中,焦虑症是广泛性焦虑症(gad)。在一实施方案中,焦虑症是恐怖症。在一实施方案中,焦虑症是应激相关障碍。在一实施方案中,提供治疗抑郁障碍、抑郁或抑郁症的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,抑郁障碍是重性抑郁。在一实施方案中,提供治疗心境障碍的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,心境障碍是快感缺乏。在一实施方案中,心境障碍是重性抑(majordepression)。在一实施方案中,提供治疗精神分裂症或情感性分裂症的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗肥胖或进食障碍的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗偏头痛的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗产后抑郁的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗与神经退行性疾病相关的心境或行为障碍的方法,该方法包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐的合成方法。在一实施方案中,提供药物组合物,其含有具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,以及至少一种药学上可接受的载体、稀释剂或赋形剂。附图简述图1解释说明ip给药(图1a)和po给药(图1b)后,代表性化合物(化合物142)在8-10周龄雄性c57bl/6j小鼠的oprk1激动剂-诱导的催乳素激发中,作为拮抗剂的活性。图2解释说明ip给药(图2a)和po给药(图2b)后,代表性化合物(化合物141)在8-10周龄雄性c57bl/6j小鼠的oprk1激动剂-诱导的催乳素激发中,作为拮抗剂的活性。图3解释说明ip给药(图3a)和po给药(图3b)后,代表性化合物(化合物145)在8-10周龄雄性c57bl/6j小鼠的oprk1激动剂-诱导的催乳素激发中,作为拮抗剂的活性。图4解释说明ip给药(图4a)和po给药(图4b)后,代表性化合物(化合物146)在8-10周龄雄性c57bl/6j小鼠的oprk1激动剂-诱导的催乳素激发中,作为拮抗剂的活性。图5解释说明ip给药(图5a)和po给药(图5b)后,代表性化合物(化合物147)在8-10周龄雄性c57bl/6j小鼠的oprk1激动剂-诱导的催乳素激发中,作为拮抗剂的活性。图6解释说明ip给药后,化合物a的酒石酸盐在8-10周龄雄性c57bl/6j小鼠的oprk1激动剂-诱导的催乳素激发中,作为拮抗剂的活性。图7解释说明在对(a)(-)-u-50,488和(b)强啡肽a响应的人oprk1上,代表性化合物(化合物142)作为有效的功能性拮抗剂的活性。相对于(c)oprm1和(d)oprd,化合物142还是选择性的。图8解释说明在利用[3h]二丙诺啡的放射配体结合试验中,相对于oprm1,代表性化合物(化合物142)作为人oprk1的有效和选择性配体的活性。图9解释说明在小鼠甩尾测试中,(-)-u-50,488(20mg/kg,i.p.)之前1h而不是24h施用的化合物142(30mg/kg,p.o.)阻断(-)-u-50,488诱导的镇痛作用。(a)潜伏期,(b)%mpr(最大可能作用=[给药后潜伏期–基线)/(15–基线)*100]。图10解释说明螺朵林(0.32mg/kg,sc;oprk1激动剂)显著增加sprague-dawley大鼠(n=8-10/组)的血浆催乳素浓度,该浓度被化合物142在第5分钟和第60分钟时间点抑制,并且被ly-2456302/cerc-501(oprk1拮抗剂)在第5分钟、第30分钟和第60分钟时间点抑制(*p≤0.05vs基线)。图11解释说明化合物142的纹状体浓度(40nm)比在颈静脉确定的浓度(~14nm)高~3倍。还测量化合物142的血浆水平(总浓度)。平均值±sem.n=4/组。图12解释说明利用雄性sprague-dawley大鼠的含有腹侧被盖区(vta)的水平脑片(~150μm)的天然组织。图13解释说明保证雄性sprague-dawley大鼠的vta放电率和/或膜电位测量的电位钳模式(vm=-60mv)。图14解释说明作为各相应细胞中产生的u69593-外向电流被化合物142(图14a)、化合物a(图14b)、pf-04455242(图14c)和ly2456302(图14d)的抑制百分数报道的剂量响应曲线。图15解释说明化合物142阻断尤其投射至中间前额皮质(mpfc)的vta多巴胺(da)神经元中的u69593响应的剂量响应曲线。图16解释说明激动剂诱导的oprm1和oprd受体对化合物142和ly2456302的剂量响应。图17解释说明(a)化合物142,(b)化合物a,(c)pf-04455242和(d)ly2456302的洗出后,基线的响应恢复。发明详述根据上文所述,本发明涉及拮抗κ阿片受体(kor)(文中还称作kor拮抗剂)的化合物,含有其的产品,及其应用和分析方法。在一实施方案中,提供具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐:其中当y是n时,x是o,或者当y是o时,x是n;r1是h、低级烷基或卤素;r2是h或低级烷基;r4和r8各自独立地是h、低级烷基、卤素或氰基,其中r4和r8中至少一个不是h或者当r4和r8都是h时,r7不是h;r5和r7各自独立地是h、卤素或氰基,其中r5和r7中至少一个是h;r6是低级烷基、低级卤代烷基、低级烷氧基、低级卤代烷氧基、环烷氧基、低级炔基、环烷基、卤素或氰基,其中当r8是低级烷基时,r6不是低级烷基;r11是–(ch2)0-1r12,其中r12是和n是1-5的整数。在一实施方案中,提供具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中当y是n时,x是o或者当y是o时,x是n;r1是h或f;r4和r8各自独立地是氢、低级烷基、卤素或氰基,其中r4和r8中至少一个不是氢或者当r4和r8都是氢时,r7不是氢;r5和r7各自独立地是氢、卤素或氰基,其中r5和r7中至少一个是h;r6是低级烷基、低级卤代烷基、低级烷氧基、低级卤代烷氧基、低级炔基、环烷基、卤素或氰基,其中当r8是低级烷基时,r6不是低级烷基;r11是–(ch2)0-1r12,其中r12是和n是1-5的整数。在另一实施方案中,提供具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中当y是n时,x是o,或者当y是o时,x是n;r1是h或f;r2是h或低级烷基;r4和r8各自独立地是氢、低级烷基、卤素或氰基,其中r4和r8中至少一个不是h或者当r4和r8都是h时,r7不是h;r5和r7各自独立地是氢、卤素或氰基,其中r5和r7中至少一个是h;r6是低级烷基、低级卤代烷基、低级烷氧基、低级卤代烷氧基、低级炔基、环烷基、卤素或氰基,其中当r8是低级烷基时,r6不是低级烷基;r11是–(ch2)0-1r12,其中r12是和n是1-5的整数。在一实施方案中,r1是h或f。如文中所用,“kor”和“oprk1”指oprk1基因编码的κ-阿片受体(kor)。“kor”和“oprk1”可在文中交互使用。如文中所用,“dor”和“oprd”指oprd基因编码的δ-阿片受体(dor)。“dor”和“oprd”可在文中交互使用。如文中所用,“mor”和“oprm1”指oprm1基因编码的μ-阿片受体(mor)。“mor”和“oprm1”可在文中交互使用。如文中所用,“低级烷基”意指具有1-8个碳原子的直链或支链烷基基团,在一些实施方案中具有1-6个碳原子,在一些实施方案中具有1-4个碳原子,并且在一些实施方案中具有1-2个碳原子。直链低级烷基基团的示例包括但不限于甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基和正辛基基团。支链低级烷基基团的示例包括但不限于异丙基、异丁基、仲丁基、叔丁基、新戊基、异戊基和2,2-二甲基丙基基团。“卤素”指氟、氯、溴和碘。“氰基”指-cn。“低级卤代烷基”指一个或多个氢原子被卤素替换的上文定义的低级烷基。低级卤代烷基基团的示例包括但不限于-cf3、-ch2cf3等。“低级烷氧基”指通过氧原子连接的上文定义的低级烷基(即-o-(低级烷基))。低级烷氧基基团的示例包括但不限于甲氧基、乙氧基、正丙氧基、正丁氧基、异丙氧基、仲丁氧基、叔丁氧基等。“低级卤代烷氧基”指通过氧原子连接的上文定义的低级卤代烷基(即-o-(低级卤代烷基))。低级卤代烷氧基基团的示例包括但不限于-ocf3、-och2cf3等。在文中,“异构体”用于包含结构的所有手性、非对映异构体或外消旋形式,除非特别指出具体的立体化学或异构形式。根据描述中显而易见的,可以任何富集程度在任何或所有不对称原子上,将所述化合物富集或拆分为光学异构体。可合成外消旋体和非对映异构体混合物,以及单一光学异构体,以便基本无其对映体或非对映异构体配偶体,并且所述都在本发明一些实施方案的范围内。由于手性中心存在产生的异构体包含一对不可重叠的异构体,称作“对映异构体”。纯化合物的单一对映异构体是光学活性的(即它们能旋转平面偏振光的平面,并指定为r或s)。“分离的光学异构体”意指已基本从相同式的相应光学异构体中纯化的化合物。例如分离的异构体可以是至少大约80%、至少80%或至少85%纯度。在其他实施方案中,分离的异构体是至少大约90重量%或至少98重量%或至少99重量%纯度。“基本对映体或非对映体”纯的意指相对于其他对映体或非对映体,一种对映体至少大约80%的对映体或非对映体富集水平,并更尤其是80%、85%、90%、95%、98%、99%、99.5%或99.9%的过量。术语“外消旋体”和“外消旋混合物”指两种对映体的等量混合物。将外消旋体标记为“(±)”,因为其不是光学活性的(即将不会以任何方向旋转平面偏振光,因为其组成的异构体彼此抵消)。“水合物”是与水分子结合存在的化合物。所述结合可包括化学计量的水,例如一水合物或二水合物或者可包括随机量的水。根据文中所用的术语,“水合物”指固体形式;也就是说,水溶液中的化合物,虽然该化合物可以是水合的,也不是文中所用的术语水合物。除了水之外的溶剂存在,“溶剂合物”类似于水合物。例如甲醇或乙醇可形成“醇合物”,醇合物还可以是化学计量的或非化学计量的。根据文中所用的术语,“溶剂合物”指固体形式;也就是说溶剂溶液中的化合物,虽然该化合物可以是溶剂合的,也不是文中所用的术语溶剂合物。“同位素”指具有相同质子数而不同中子数的原子,并且式(i)化合物的同位素包括任何此类化合物,其中一个或多个原子被所述原子的同位素替换。例如,最常见的碳形式碳12具有6个质子和6个中子,而碳13具有6个质子和7个中子,并且碳14具有6个质子和8个中子。氢具有两个稳定的同位素氘(1个质子和1个中子)和氚(1个质子和2个中子)。并且,氟具有许多同位素,氟19是寿命最长的。因此,具有式(i)结构的化合物的同位素包括但不限于式(i)化合物,其中1个或多个碳12原子被碳13和/或碳14原子替换,其中1个或多个氢原子被氘和/或氚替换,和/或其中1个或多个氟原子被氟19替换。“盐”通常指与反荷离子结合的离子形式的有机化合物例如羧酸或胺。例如,阴离子形式的酸和阳离子形成的盐称作“酸加成盐”。相反地,阳离子形式的碱和阴离子形成的盐称作“碱加成盐”。具有式(i)结构的化合物的共晶形式还包括在本发明范围内;即两种或更多种不同分子和/或离子化合物通常以化学计量比率组成晶状单相物质的固体,其即不是溶剂合物,也不是简单的盐。术语“药学上可接受的”指批准用于人消费且通常无毒的物质。例如,术语“药学上可接受的盐”指无毒的无机或有机酸和/或碱加成盐(参见例如lit等人,saltselectionforbasicdrugs,intj.pharm.,33,201-217,1986)(引入文中作为参考)。本发明化合物药学上可接受的碱加成盐包括例如金属盐包括碱金属盐、碱土金属盐和过渡金属盐例如钙盐、镁盐、钾盐、钠盐和锌盐。药学上可接受的碱加成盐还包括由碱性胺制备的有机盐例如n,n'-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、葡甲胺(n-甲基葡糖胺)和普鲁卡因。可由无机酸或有机酸制备药学上可接受的酸加成盐。无机酸的示例包括盐酸、氢溴酸、氢碘酸、硝酸、碳酸、硫酸和磷酸。适当的有机酸可选自脂肪族、环脂肪族、芳香族、芳香族脂肪族、杂环族的羧酸和磺酸类有机酸,其示例包括甲酸、乙酸、丙酸、琥珀酸、羟基乙酸、葡糖酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、葡萄糖醛酸、马来酸、富马酸、丙酮酸、天冬氨酸、谷氨酸、苯甲酸、邻氨基苯甲酸、4-羟基苯甲酸、苯乙酸、苦杏仁酸、马尿酸、丙二酸、草酸、双羟萘酸(扑酸)、甲磺酸、乙磺酸、苯磺酸、泛酸、三氟甲磺酸、2-羟基乙磺酸、对甲苯磺酸、对氨基苯磺酸、环己胺基磺酸、硬脂酸、海藻酸、β-羟基丁酸、水杨酸、粘酸和半乳糖醛酸。尽管药学上不可接受的盐通常不可用作药物,所述盐可用作例如具有式i结构的化合物合成中的(例如通过重结晶纯化它们)的中间体。在更具体的实施方案中,提供具有式(ii)至(xx)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐:在另外的实施方案中,r2是h,并提供具有式(i-a)至(xx-a)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐。在另外的实施方案中,r1是h,并提供具有式(i-b)至(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐。在更具体的实施方案中,提供具有式(r,r)-(xviii-a)、(s,r)-(xviii-a)、(r,s)-(xviii-a)、(s,s)-(xviii-a)中任何一式结构的式(xviii-a)化合物的异构体或其药学上可接受的水合物、溶剂合物、同位素或盐:在更具体的实施方案中,提供具有式(r,r)-(xix-a)、(s,r)-(xix-a)、(r,s)-(xix-a)、(s,s)-(xix-a)中任何一式结构的式(xix-a)化合物的异构体或其药学上可接受的水合物、溶剂合物、同位素或盐:在更具体的实施方案中,提供具有式(r,r)-(xx-a)、(s,r)-(xx-a)、(r,s)-(xx-a)、(s,s)-(xx-a)中任何一式结构的式(xx-a)化合物的异构体或其药学上可接受的水合物、溶剂合物、同位素或盐:在更具体的实施方案中,提供具有式(r,r)-(xviii-b)、(s,r)-(xviii-b)、(r,s)-(xviii-b)、(s,s)-(xviii-b)中任何一式结构的式(xviii-b)化合物的异构体或其药学上可接受的水合物、溶剂合物、同位素或盐:在更具体的实施方案中,提供具有式(r,r)-(xix-b)、(s,r)-(xix-b)、(r,s)-(xix-b)、(s,s)-(xix-b)中任何一式结构的式(xix-b)化合物的异构体或其药学上可接受的水合物、溶剂合物、同位素或盐:在更具体的实施方案中,提供具有式(r,r)-(xx-b)、(s,r)-(xx-b)、(r,s)-(xx-b)、(s,s)-(xx-b)中任何一式结构的式(xx-b)化合物的异构体或其药学上可接受的水合物、溶剂合物、同位素或盐:在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是低级烷基、卤素或氰基,且r8是h。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是低级烷基,且r8是h,并且更具体地其中r4是甲基且r8是h。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是卤素,且r8是h,并且更具体地其中r4是f或cl且r8是h。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是氰基,且r8是h。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是氰基,且r8是低级烷基、卤素或氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是h,且r8是低级烷基,并且更尤其是其中r4是h且r8是甲基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是h,且r8是卤素,并且更尤其是其中r4是h且r8是f或cl。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是h,且r8是氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4和r8各自独立地是低级烷基、卤素或氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4和r8各自独立地是甲基、f、cl或氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是甲基且r8是f、cl或氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r4是f、cl或氰基且r8是甲基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r5和r7都是h。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r5是h,且r7是f或氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r5是卤素或氰基,且r7是h,并且更尤其是r5是f或氰基,且r7是h。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r6是低级烷基,并更尤其是r6是甲基或乙基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r6是低级烷基,并更尤其是r6是–ch2cf3。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r6是环丙基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r6是低级烷氧基,并更尤其是r6是甲氧基。在其他实施方案中,提供具有式(i)~(xx)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r2是低级烷基,并更尤其是甲基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中r11是且更尤其是r11是在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中:r4是h;r5是h;r6是低级烷基,并更尤其是甲基或乙基;r7是h;r8是卤素,并更尤其是f或cl。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中:r4是低级烷基,并更尤其是甲基;r5是h;r6是低级烷基,并更尤其是甲基或乙基;r7是h;r8是卤素,并更尤其是f或cl。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中:r4是低级烷基,并更尤其是甲基;r5是h;r6是低级烷基,并更尤其是甲基或乙基;r7是h;r8是氰基。在其他实施方案中,提供具有式(i)~(xx)、式(i-a)~(xx-a)、式(i-b)~(xx-b)中任何一式结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐,其中:r4是氰基;r5是h;r6是低级烷基,并更尤其是甲基或乙基;r7是h;r8是h或卤素。在更具体的实施方案中,提供具有式(ii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐:其中:r1是h或f;r2是h或低级烷基;r4是h、ch3、cl或cn;r5是h或f;r6是cl、br、f、cn、ch3、cf3、ch2ch3、chfch3、cf2ch3、ch2ch2f、ch2cf3、ch(ch3)2、c≡cch3、环丙基、环丁基、och3、ocf3、och2ch3或och(ch3)2;r7是h或f;r8是h、f、cl或cn;r11是–(ch2)0-1r12,其中r12是其中r5和r7中至少一个是h;当r2是h时,r4和r8中至少一个不是h或者当r2、r4和r8是h时,r7不是h;并且当r8是低级烷基时,r6不是低级烷基。在更具体的实施方案中,提供具有式(x)结构的化合物或其其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐:其中:r1是h或f;r2是h或低级烷基;r4是h、ch3、cl或cn;r5是h或f;r6是cl、br、f、cn、ch3、cf3、ch2ch3、chfch3、cf2ch3、ch2ch2f、ch2cf3、ch(ch3)2、c≡cch3、环丙基、环丁基、och3、ocf3、och2ch3或och(ch3)2;r7是h或f;r8是h、f、cl或cn;r11是–(ch2)0-1r12,其中r12是其中r5和r7中至少一个是h;当r2是h时,r4和r8中至少一个不是h,或者当r2、r4和r8是h时,r7不是h;并且当r8是低级烷基时,r6不是低级烷基。代表性化合物包括表1中所列的化合物,及其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐。表1代表性化合物在一些实施方案中,本发明提供药物组合物,其含有本发明化合物,以及至少一种药学上可接受的载体、稀释剂或赋形剂。例如,通常将活性化合物与载体混合或者用载体稀释或者包封在载体(其可以是以安瓿、胶囊、囊袋(sachet)、纸或其他容器)中。当将活性化合物与载体混合时或者当将载体作为稀释剂时,载体可以是固体、半固体或液体材料,作为活性化合物的溶媒、赋形剂或基质。可将活性化合物吸附在粒状固体载体上,例如包含于囊袋中。适当载体的一些示例是水、盐溶液、醇类、聚乙二醇类、聚羟基乙氧基化蓖麻油(polyhydroxyethoxylatedcastoroil)、花生油、橄榄油、明胶、乳糖、白土、蔗糖、糊精、碳酸镁、糖、环糊精、直链淀粉、硬脂酸镁、滑石、明胶、琼脂、果胶、阿拉伯胶、硬脂酸或纤维素的低级烷基醚、硅酸、脂肪酸类、脂肪酸胺类、脂肪酸单甘油酯类和脂肪酸二甘油酯类、脂肪酸季戊四醇酯类、聚氧乙烯、羟甲基纤维素和聚乙烯吡咯烷酮。类似地,载体或稀释剂可包括任何本领域已知的缓释材料例如单硬脂酸甘油酯或二硬脂酸甘油酯(其是单独的或者与蜡混合的)。制剂可与不与活性化合物发生不利反应的辅助物质混合。所述添加剂可包括润湿剂、乳化剂和混悬剂、影响渗透压的盐、缓冲剂和/或着色剂、防腐剂、甜味剂或矫味剂。如果需要,还可以将组合物灭菌。施用途径可以是将本发明的活性化合物有效转运至适当或需要作用位置的任何途径,例如口服、经鼻腔、经肺、含服、经皮下、经真皮、透皮或肠胃外的途径,诸如直肠、储库、皮下、静脉内、尿道内、肌内、鼻内、眼用溶液剂或软膏剂,口服途径是优选的。对于肠胃外施用,载体将通常包含无菌水,尽管还可包括辅助溶解或作为防腐剂的其他成分。此外,还可制备注射用混悬剂,其中可利用适当的液体载体、助悬剂等。对于局部施用,可利用温和、保湿基质例如软膏或乳膏配制本发明化合物。如果将固体载体用于口服施用,可将制剂制片,将制剂以粉末或小丸形式置于硬明胶胶囊中或制剂可以是含片(troche)或糖锭剂(lozenge)形式。如果使用液体载体,制剂可以是糖浆剂、乳化剂、软明胶胶囊剂或无菌注射液体例如水性或无水液体混悬剂或溶液剂形式的。注射剂型通常包括水性混悬剂或油性混悬剂,其可利用适当的分散剂或润湿剂和助悬剂制备。注射形式可以是在溶液相中或者以混悬液形式,其用溶剂或稀释剂制备。可接受的溶剂或溶媒包括无菌水、林格(ringer)溶液或等渗水性盐溶液。或者,可将无菌油用作溶剂或助悬剂。优选地,油或脂肪酸是非挥发性的,包括天然或合成油类,脂肪酸类,单-、二-或三-甘油酯类。对于注射,制剂还可以是适合于用上文所述的适当溶剂重构的粉末。所述示例包括但不限于冻干、旋干或喷雾干燥的粉末、无定型粉末、颗粒剂、沉淀物或微粒(particulates)。对于注射,制剂可任选地包含稳定剂、ph调节剂、表面活性剂、生物利用度调节剂及其组合。可将化合物配制用于通过注射而肠胃外施用,例如通过单次快速静脉注射或连续输注。用于注射的单位剂型可以是在安瓿瓶或多剂量容器中。可将本发明制剂设计,以便利用本领域众所周知的方法施用至患者后,提供活性成分的快速、持久或延迟释放。因此,还可配制制剂用于控释或缓释。本发明考虑的组合物可包括例如胶束或脂质体或一些其他的包囊形式,或者可以以延长释放形式施用,以提供延长的贮存和/或递送效果。因此,可将制剂压制成丸剂或筒体,并将其肌内或皮下植入,作为储库注射剂(depotinjections)。所述植入物可使用已知的惰性材料例如聚硅酮类和可生物降解多聚体例如聚丙交酯-聚乙交酯。其他可生物降解多聚体的示例包括聚(原酸酯)和聚(酐)。对于鼻施用,制剂可包含溶解或悬浮于液体载体优选水性载体中的本发明化合物,用于气雾剂应用。载体可包含添加剂例如增溶剂诸如丙二醇、表面活性剂、吸收促进剂如卵磷脂(磷脂酰胆碱)或环糊精或者防腐剂如对羟苯甲酸酯类。对于肠胃外施用,特别适合的是注射溶液剂或混悬剂,优选含有溶解于多羟基化蓖麻油中的活性化合物的水性溶液剂。可将剂型每天一次或者每天多于一次,例如每天两次或三次施用。或者,如果处方医生发现是合理的,那么可将剂型比每天更低的频率,例如每两天或每周施用。给药方案包括例如剂量调整至所需程度或要治疗适应症可用的程度,因此保证患者身体适合所述治疗和/或将与治疗相关的不需要的副作用最小化或避免与治疗相关的不需要的副作用。其他剂型包括延迟释放或控释形式。适当的施用方案和/或剂型包括例如physicians'deskreferenc最后一版中提出的那些,将其引入文中作为参考。当用于预防疾病或病症发作时,通常根据医生的建议或在医生监督下,将文中提供的化合物以上文所述的剂量水平施用至具有患所述病症风险的个体。具有患特定疾病或病症风险的个体通常包括具有该疾病家族史的那些或者已通过基因测定或筛选确定特别易患该疾病或病症的那些。长期施用指化合物或其药物组合物的施用历经一段长期的时间例如历经3个月、6个月、1年、2年、3年、5年等或者可无限期地持续例如个体余下的生命期。在一些实施方案中,长期施用预期在血液中提供恒定水平的化合物,例如在长期的治疗窗内。在另一实施方案中,提供制备文中所述化合物的组合物的方法,其包括用药学上可接受的载体或稀释剂配制本发明的化合物。在一些实施方案中,药学上可接受的载体或稀释剂适合于口服施用。在一些所述实施方案中,所述方法可另外包括将组合物配置成片剂或胶囊剂的步骤。在其他实施方案中,药学上可接受的载体或稀释剂适合于肠胃外施用。在一些所述实施方案中,方法还包括冻干组合物,以形成冻干制剂的步骤。在另一实施方案中,提供拮抗kor的方法,该方法包含使受体与有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物接触。文中,术语“拮抗”用于涵盖以一定方式与受体相互作用并由此通过在其天然配体的结合位置或结合位置之外的位置与受体结合而起拮抗剂作用的分子。“κ阿片受体”或“kor”是阿片受体家族的一员,作为主要内源性配体,其结合阿片样肽强啡肽。文中所用的短语“kor拮抗”涵盖以一定方式与kor相互作用并由此通过在强啡肽位置或者在除该结合位置之外的位置(即变构结合)结合至kor而起拮抗剂作用的分子。在一实施方案中,提供治疗神经精神病学病患或行为病患的方法,无论是器质性的、应激诱导的或医源性的,其特征在于血清催乳素升高并对kor拮抗剂施用响应而伴随血清催乳素降低。所述方法包含向个体以足以为个体提供有益作用的频率和持续期施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在另一实施方案中,提供在医学上指示的kor拮抗的个体中治疗不良状况的方法。所述方法包含以足以为个体提供有益作用的频率和持续期向个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。根据文中所用,“个体”意指哺乳动物或非哺乳动物。哺乳动物包括例如人;非人的灵长类例如猿类和猴类;牛;马;绵羊;和山羊。非哺乳动物动物包括例如鱼和鸟类。文中意思范围内的“治疗”指与病患或疾病相关的症状的减轻,或者抑制所述症状的进一步增进或恶化,或者在一定条件下预防疾病或病症。当用于描述本发明化合物在为患kor介导的病症或不良状况的个体提供治疗中的用途时,表述“有效量”指本发明化合物作为拮抗剂,有效结合至个体组织中的kor的量,其中kor与病症相关,其中所述结合发生至足够在个体中产生有益治疗作用的程度。术语“不良状况”用于描述任何疾病、病患或病症,并可交互使用,并且在本申请的上下文中,指kor在与不良状况或其症状相关的生化机制中起作用的疾病、病患或病症,以便可通过作用在所述kor上而实现治疗上的有益作用。在一些实施方案中,本发明提供用本发明化合物拮抗kor的方法。所述方法涉及使受体与适当浓度的化合物接触,以拮抗受体。接触可发生在体外例如完成如下测试中,即进行与监管批准的申报相关的试验以测定本发明化合物的kor抑制活性的测试。在一些实施方案中,拮抗kor的方法还可以在体内进行,也就是说,在哺乳动物例如人患者或受试动物(文中称作“个体”)活体内。可通过上文所述途径之一例如口服,将本发明化合物提供至活的有机体或者可将本发明化合物局部提供在身体组织中。在本发明化合物存在下,发生受体的抑制,并且可研究其作用。本发明提供的治疗方法包括本发明化合物单独施用或与另一种药理学活性物质或第二种药物组合施用至个体或者具有不良状况的患者,对于其,医学上指示拮抗kor,例如成瘾症包括与物质滥用或成瘾相关的病症;焦虑症;抑郁障碍;心境障碍;精神分裂症或情感性分裂症;应激相关障碍;肥胖和进食障碍;偏头痛;产后抑郁;神经变性疾病或病症,包括与神经变性疾病相关的心境和行为障碍;产后抑郁;麻醉和/或镇静;癫痫;癫痫持续状态;和发作(seizure)。在一实施方案中,提供治疗成瘾症包括与物质滥用或成瘾相关的病症和强迫行为的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。文中所述的与物质滥用或成瘾相关的病症可包括赌博、药物成瘾、药物滥用、酒精依赖、酒精滥用、物质诱导的抑郁,以及物质例如酒精、尼古丁、苯丙胺、甲基苯丙胺、可卡因、阿片成瘾、海洛因成瘾、苯二氮类等诱导的心境障碍。在一实施方案中,提供治疗cns-相关病症的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。cns-相关病症包括物质滥用相关的病症和/或戒断综合征、心境障碍、焦虑症、精神分裂症谱系障碍(schizophreniaspectrumdisorders)、疼痛、人格障碍、孤独症谱系障碍、进食障碍;睡眠障碍;记忆和/或认知障碍、头部冲击和创伤性脑损伤;血管性疾病和认知障碍。示例性cns病患包括物质滥用病症和/或戒断综合征(包括阿片类、可卡因和/或酒精成瘾);心境障碍(包括抑郁、心境恶劣障碍、双相情感障碍);焦虑症,以及包括强迫障碍例如强迫症(obsessive-compulsivedisorder,ocd),社交恐怖症,广泛性焦虑症(gad),社交焦虑症;应激、创伤后应激障碍(ptsd);精神分裂症谱系障碍(包括精神分裂症、情感性分裂症);疼痛(包括偏头痛、神经性疼痛、伤害相关疼痛综合征、急性疼痛、慢性疼痛);人格障碍(包括反社会型人格障碍、强迫性人格障碍);孤独症谱系障碍(asd)(包括孤独症,单成因性孤独症例如synaptophathy's,如rett综合征、脆性x综合征、angelman综合征);进食障碍;睡眠障碍(包括失眠症);记忆和/或认知障碍(包括注意力障碍(例如注意力缺陷多动障碍(adhd)),痴呆(包括阿尔茨海默氏型痴呆、路易斯体型痴呆、血管型痴呆),头部冲击和创伤性脑损伤(tbi);血管性疾病(包括中风、缺血、血管畸形)和认知障碍(包括阿尔茨海默氏病和其他形式痴呆)。在一实施方案中,提供治疗焦虑症的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。焦虑症是覆盖几种不同类型异常和病态恐惧与焦虑的总称。现行精神病诊断标准识别多种焦虑症,包括广泛性焦虑症、惊恐障碍、应激相关障碍、强迫症、恐怖症、社交焦虑症、分离焦虑症和创伤后应激障碍(ptsd)。在一实施方案中,焦虑症是社交焦虑症。在一实施方案中,焦虑症是恐怖症。在一实施方案中,焦虑症是应激相关障碍。在一实施方案中,焦虑相关病症是ptsd。广泛性焦虑症是以长期焦虑为特征的常见慢性病症,其不集中于任何一个个体或情形。患广泛性焦虑症的人遭受非特异性持续的恐惧和担忧,并对日常事务变得过度关切。广泛性焦虑症是最常见的影响成年人的焦虑症。惊恐障碍中,人遭受强烈恐惧和忧虑的短暂发作,通常表示为震颤、晃动、意识错乱、眩晕、恶心、呼吸困难。通过apa定义为恐惧或不适(突然出现并在不到10分钟内达到峰值)的这些惊恐发作可持续几小时,并可由应激、害怕或者甚至锻炼引发;尽管具体原因不总是显而易见的。除了再出现的出乎意料的惊恐发作,惊恐障碍的诊断还要求所述发作具有长期后果:过度担忧发作潜在的牵涉,未来发作的持续恐惧或者与发作相关行为的显著变化。因此,患惊恐障碍的患者经历甚至超出具体的惊恐发作的症状。通常,惊恐患者注意到心跳的正常变化,会致使他们认为心脏出现问题或者他们要经历另一场惊恐发作。在一些情况下,躯体功能增高的警觉(过度警觉)发生在惊恐发作期间,其中将任何感觉到的生理变化理解为可能威胁生命的疾病(即过度臆想症)。强迫症是主要以反复强迫思维(痛苦的、固执的和侵入性思想或想象)和强迫(迫切完成具体动作或仪式)为特征的一类焦虑症。ocd思维模式可以与迷信相比,因为其涉及对实际不存在的因果关系的信仰。通常,过程是完全无逻辑的;例如,以某种姿势走路的强迫可用于减轻即将来临的伤害的固执念头。并在许多情况下,强迫是完全不能理解的,只是迫切完成神经过敏引发的惯例。在少数情况下,ocd患者可仅经历无明显强迫的强迫观念;更少数的患者仅经历强迫。一种最大类型的焦虑症是恐怖症,其包括具体刺激或情形引发的恐惧和焦虑的所有情况。患者通常从遇到其恐惧的目标预期恐怖后果,其可以是从社交恐怖症、特定恐怖症、广场恐怖症、动物至位置至体液的恐怖症中的任何一种。创伤后应激障碍或ptsd是由创伤性经历引起的焦虑症。创伤后应激可由极端情形引起,例如战斗、强奸、人质处境或者甚至严重的事故。其还可以由长期(慢性的)暴露于严重应激源而引起,例如经历个别战斗但不能处理连续斗争的战士。常见症状包括后退、回避行为和抑郁。在一实施方案中,提供治疗抑郁障碍、抑郁或抑郁疾患的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。所述病症的示例包括重性抑郁、抗药性抑郁、心境恶劣和双相情感障碍。在一实施方案中,提供治疗心境障碍或情感性障碍的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。心境障碍或情感性障碍包括重性抑郁障碍(mdd);双相情感障碍;快感缺乏;心境恶劣;重性抑郁、精神重性抑郁(pmd)或精神病性抑郁;产后抑郁;季节性情感性障碍(sad);并且紧张性精神抑郁是一种罕见且严重形式的重性抑郁,与运动行为和其他症状的失调相关。术语“快感缺乏”和“快感缺乏症状”可交换使用,并将其定义为不能从通常可寻找快乐的活动例如锻炼、嗜好、音乐、性活动或社交中感受快乐。术语“快感缺乏”和“快感缺乏症状”与dsm-5中定义为忧郁性抑郁的“具有忧郁特征的抑郁障碍”的标准紧密相关,所述忧郁性抑郁以大多数或所有活动中的乐趣丧失、对快乐刺激反应失败、比悲痛或失去更显著的抑郁心境的质量、早上时刻的症状恶化、清早醒来、精神运动性阻抑、体重过度减轻或过度内疚为特征。术语“具有忧郁特征的抑郁障碍的治疗”包括抑郁障碍和与其相关的忧郁特征的治疗。在一实施方案中,心境障碍是快感缺乏。在一实施方案中,心境障碍是重性抑郁。在一实施方案中,心境障碍是季节性情感性障碍(sad)。在其他实施方案中,提供治疗精神分裂症或情感性分裂症的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在其他实施方案中,提供治疗肥胖或进食障碍的方法,该方法包括以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。文中所述的肥胖和进食障碍可包括食欲亢进、神经性厌食等。在其他实施方案中,提供治疗偏头痛的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在另一实施方案中,预防性治疗是提供预防偏头痛。在该方面,将kor拮抗推荐为具有患偏头痛风险的个体中偏头痛的预防治疗。在一实施方案中,提供治疗产后抑郁的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。出生后,孕酮水平立即显著降低,引起产后抑郁(pnd)的发生。pnd的症状是轻度抑郁至需要住院治疗的精神病。pnd还与严重焦虑和易怒相关。pnd-相关的抑郁不遵从经典抗抑郁的治疗,并且经历pnd的妇女显示经前期综合征(pms)发病率增加。在其他实施方案中,提供治疗神经变性疾病或病症的方法,所述神经变性疾病或病症包括与神经变性疾病相关的心境和行为障碍;麻醉和/或镇静;癫痫;发作(seizure);糖尿病、糖尿病并发症、糖尿病视网膜病变;性/生殖障碍;高血压;脑出血;充血性心力衰竭;动脉粥样硬化;类风湿性关节炎;高脂血症、三酸甘油酯过高症、高血糖症、高脂蛋白血症;强迫行为障碍(例如狗的舔爪子)和脊柱损伤,所述方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。在一实施方案中,提供治疗与神经变性疾病或病症相关的心境和行为障碍的方法,该方法包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。术语“神经变性疾病”包括与神经元结构或功能进行性丧失或者神经元死亡相关的疾病和病症。神经变性疾病和病症包括但不限于阿尔茨海默氏病(包括轻度、中度或严重认知损害相关的症状);肌萎缩侧索硬化(als);缺氧和缺血性损伤;共济失调和惊厥(包括用于治疗和预防及预防情感性分裂症或者用于治疗精神分裂症的药物引起的发作);良性健忘;脑水肿;小脑性共济失调包括mcleod神经性棘红细胞增多综合征(mls);闭合性头部损伤;昏迷;挫伤性损伤(例如脊髓损伤和头损伤);痴呆包括多发性梗死痴呆和老年痴呆;意识障碍;唐氏(down)综合征;药物引起的或药物治疗引起的帕金森症(例如精神安定剂诱发的急性静坐不能、急性肌张力障碍、帕金森症或迟发性运动障碍、抗精神病药致恶性综合征或药疗法诱发的体位震颤);癫痫;脆性x综合征;吉累斯·德拉图雷特(gillesdelatourette's)综合征;头损伤;听力损害和丧失;亨廷顿病;伦诺克斯(lennox)综合征;左旋多巴诱导运动障碍;精神发育迟滞;运动障碍包括运动不能(akinesias)和运动不能(僵硬)综合征(包括基底神经节钙化、皮质基底变性、多系统萎缩、帕金森症-als痴呆复合体、帕金森氏病、脑炎后帕金森症和进行性核上麻痹);肌肉痉挛和与肌痉挛状态或虚弱相关的病症包括舞蹈病(例如良性遗传性舞蹈症、药物所致舞蹈症、偏身颤搐、亨廷顿病、神经棘红细胞增多症、西登哈(sydenham's)舞蹈病和症状性舞蹈病),运动障碍(包括抽搐例如复杂性抽搐、简单性抽搐和症状性抽搐),肌阵挛(包括全身肌阵挛和病灶肌阵挛),震颤(例如休息性震颤、姿势性震颤和意向性震颤)和肌张力障碍(包括轴向肌张力障碍(axialdystonia)、张力障碍书写痉挛、偏瘫性肌张力障碍、突发性肌张力障碍和病灶性张力障碍诸如眼睑痉挛、口下颌肌张力障碍和痉挛性发音困难和斜颈);神经元损害包括眼睛损伤、眼睛视网膜病变或黄斑变性;脑中风、血栓栓塞中风、出血性中风、脑缺血、脑血管痉挛、低血糖症、健忘症、缺氧、缺氧症、围产期窒息和心搏骤停后的神经中毒性损伤;帕金森氏病;发作;statusepilecticus;中风;耳鸣;管状硬化症和病毒感染诱导的神经变性(例如获得性免疫缺陷综合征(aids)和脑病诱导的)。神经变性疾病还包括但不限于脑中风、血栓栓塞中风、出血性中风、脑缺血、脑血管痉挛、低血糖症、健忘症、缺氧、缺氧症、围产期窒息和心搏骤停后的神经毒性损伤。治疗或预防神经变性疾病的方法还包括治疗或预防作为神经变性疾病特征的神经元功能丧失。在一实施方案中,提供麻醉和/或镇静方法,其包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。麻醉是药理学上诱导的且可逆的记忆缺失、镇痛、反应性丧失、骨骼肌反射丧失、应激反应降低或所述全部同时发生的状态。所述作用可由单一药物获得,该药物单独提供作用的正确组合或者有时与药物组合(例如安眠药、镇静药、麻痹药、镇痛药),以实现非常具体的组合结果。麻醉保证患者在无患者否则应经历的应激和疼痛条件下,进行手术和其他操作。镇静是通过施用药物而减轻易怒或激动,通常以便于医学操作或诊断操作。镇静和镇痛包括最小镇静(焦虑解除)至全身麻醉的意识状态连续谱。最小镇静还称作焦虑解除。最小镇静是患者对口头命令正常反应的药物诱导状态。可损害认知功能和协调性。呼吸和心血管功能通常不受影响。中度镇静/镇痛(连续镇静)是药物诱导的意识抑制,期间患者对单独或者伴随光触觉刺激的口头命令有目的地响应。通常不需要干预,以维持气道通畅。自发通气一般是足够的。通常维持心血管功能。深度镇静/镇痛是药物诱导的意识抑制,期间患者不能容易唤醒但重复或疼痛刺激后有目的的响应(无疼痛刺激的反射撤回)。可损害独立的通气功能,并且患者可能需要辅助来维持气道通畅。自发换气可能是不足够的。通常维持心血管功能。全身麻醉是药物诱导的意识丧失,期间患者甚至对疼痛刺激不是觉醒的。通常损害维持独立通气功能的能力,并且通常需要辅助,以维持气道通畅。由于受抑的自发通气或药物诱导的神经肌肉功能抑制,可能需要正压通气。可损害心血管功能。重症监护病房(icu)中的镇静保证患者环境意识的抑制和对外部刺激的响应的降低。其可在危重病症者的护理中起作用,并包括患者及其疾病过程中个体间将变化的广谱症状控制。已将危重护理中的重度镇静用于促进外插导气管(endrotrachealtube)耐受和呼吸机同步,时常用神经肌肉阻滞剂。在icu中,诱导镇静期(例如长期镇静、持续镇静),并维持延长的一段时间(例如1天、2天、3天、5天、1周、2周、3周、1个月、2个月)。长期镇静物质可具有长期作用。icu中的镇静物质可具有短的清除半衰期。程序化镇静和镇痛还称作连续镇静,是在镇痛或不镇痛的条件下,施用镇静药或解离剂的技术,以诱导保证个体耐受不愉快的操作并维持心肺功能的状态。在一实施方案中,提供治疗癫痫的方法,其包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。癫痫是特征为随时间推移重复发作的脑部病症。癫痫的类型可包括但不限于全身性癫痫例如儿童失神性癫痫、青少年期肌阵挛性癫痫、醒时癫痫大发作性癫(epilepsywithgrand-malseizuresonawakening)、west综合征、lennox-gastaut综合征,局限性癫痫(partialepilepsy)例如颞叶癫痫、额叶癫痫、儿童良性局灶性癫痫。在一实施方案中,提供治疗癫痫持续状态的方法,其包括以足以为个体提供有益作用的频率和持续期,向需要其的个体施用有效量的具有式(i)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物,。癫痫持续状态(se)可包括例如惊厥性癫痫持续状态例如早期癫痫持续状态、已确立(established)癫痫持续状态、顽固性癫痫持续状态、超顽固性癫痫持续状态;非惊厥性癫痫持续状态例如全身性癫痫持续状态、复杂部分型癫痫持续状态;全身周期性癫痫样放电;和周期性单侧癫痫样放电。惊厥性癫痫持续状态是以存在惊厥性癫痫持续状态发作为特征,并可包括早期癫痫持续状态、已确立癫痫持续状态、顽固性癫痫持续状态、超顽固性癫痫持续状态。早期癫痫持续状态用一线疗法治疗。已确立癫痫持续状态以癫痫持续状态发作为特征,尽管用一线疗法治疗,其仍持续,并且施用二线疗法。顽固性癫痫持续状态是以癫痫持续状态发作为特征,尽管用一线疗法和二线疗法治疗,其仍持续,并且通常施用全身麻醉药。超顽固性癫痫持续状态是以癫痫持续状态发作为特征,尽管用一线疗法,其仍持续二线疗法治疗,和全身麻醉药持续24小时或更长时间。非惊厥性癫痫持续状态可包括例如局部非惊厥性癫痫持续状态例如复杂部分型非惊厥性癫痫持续状态、简单部分型非惊厥性癫痫持续状态、精细型(subtle)非惊厥性癫痫持续状态;全身型非惊厥性癫痫持续状态例如迟发失神型非惊厥性癫痫持续状态、非典型失神型非惊厥性癫痫持续状态或者典型失神型非惊厥性癫痫持续状态。在一实施方案中,提供治疗发作(seizure)的方法,其包含以足以为个体提供有益作用的频率和持续期向需要其的个体施用有效量的具有式(i)~(xvii)结构的化合物或其药学上可接受的异构体、外消旋体、水合物、溶剂合物、同位素或盐或者含有其的药物组合物。发作是脑部异常电活动发作后发生的身体结果或行为变化。术语“发作”时常与“惊厥”交换使用。惊厥是人体快速且不受控的震颤。惊厥期间,人的肌肉反复收缩和松弛。基于行为和脑活动的类型,将发作分成两大类:全身的和部分的(还称作局部的或局灶的)。发作方式分类帮助医生诊断是否患者患有癫痫。遍布全脑的电脉冲产生全身发作,而相对小部分脑的电脉冲产生(至少初始)部分发作。产生发作的脑部分有时称作病灶。有6种类型的全身发作。最常见且剧烈的,并由此最众所周知的是全身性惊厥,也称作癫痫大发作。在该类型的发作中,患者失去意识,并通常崩溃。意识丧失后是全身僵硬30~60秒(称作“发作”的紧张期),然后猛烈急动30~60秒(称作“阵挛”期),随后患者进入深度睡眠(发作后阶段)。癫痫大发作期间,可发生伤害和事故,例如咬舌和尿失禁。失神型发作引起意识的短暂丧失(仅几秒),具有少数症状或无症状。患者(最常见儿童)通常中断活动并茫然地凝视。所述发作迅速开始和结束并可一天发生数次。患者通常未意识到他们正在发作,除了他们可能意识到“损失时间”。肌阵挛性癫痫发作由散发的急动(jerk)构成,通常在身体的两侧。患者有时将急动描述为短暂的电休克。当剧烈时,所述发作可导致跛行步态或不由自主地丢物体。阵挛性发作是重复性、有节律的急动,其同时涉及身体两侧。张力性发作,以肌肉僵硬为特征。无张力性癫痫发作由特别是胳膊和腿部肌张力的突然和全身丧失构成,其经常造成摔倒。文中所述的发作可包括癫痫性发作;急性重复性发作;丛集型发作;连续发作;持续发作;长时间发作;反复发作;癫痫持续状态发作例如顽固型惊厥性癫痫持续状态、非惊厥性癫痫持续状态发作;顽固性发作;肌阵挛性发作;张力性发作;张力性-阵挛性癫痫发作;简单部分发作;复杂部分发作;继发性全身性癫痫发作;非典型失神型发作;失神型发作;无张力性癫痫发作;良性罗兰样(rolandic)发作;发热性发作;情绪性发作;病灶性发作;痴笑型发作;全身发作性癫痫发作;婴儿痉挛;杰克逊(jacksonian)发作;双侧大量肌痉挛发作;多病灶发作;新生儿发作性癫痫;夜间癫痫发作;枕叶癫痫发作;损伤后癫痫发作;精细性发作;sylvan发作;视反射发作;或者撤药发作。可利用本领域技术人员已知的标准合成技术,合成具有式(i)结构、以及式(ii)~(xvii)亚结构的化合物。例如,可利用流程图1和2提出的通用合成方法,合成本发明化合物。为了这个目的,文中所述的反应、过程和合成方法不限于下述实验部分所述的具体条件,而是预期作为本领域技术人员的指导。例如,可在任何适当的溶剂或其他试剂中,进行反应,以完成所需转化。一般来讲,适当的溶剂是质子或非质子溶剂,在进行反应的温度(即可以是冰冻至沸腾温度范围内的温度)下,其是与反应物、中间体或产物基本上不反应的。可在一种溶剂或多于一种溶剂的混合物中,完成指定反应。根据具体的反应,可以应用反应后具体后处理的适当溶剂。流程图1试剂和条件:i)a(1.0当量(equiv.)),b(1.2当量),pocl3,110℃,1h;ii)1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2当量),dipea(1.2当量),etoh,120℃,18h,33%(通过两步);iii)10%aq.h2so4,thf,45℃,2h,定量产率;iv)胺(2.0当量),nabh(oac)3(3.0当量),acoh(3.0当量),dipea(2.0当量),1,2-二氯乙烷(dce),室温(rt),18h,93%产率。流程图2试剂和条件:i)dmf(2.5当量),pocl3(7.0当量),0至75℃,48h,36-39%;ii)nah2po4(5.0当量),naclo2(3.0当量),na2so3(4.0当量),ch3cn,94-98%;iii)a)15(1.0当量),socl2(3.0当量),ch2cl2,dmf(催化的),50℃,2h,b)乙酰胺肟(1.2当量),dipea(1.2当量),二氧六环,100℃,4h,48-54%;iv)1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2当量),dipea(2.0当量),etoh,125℃,过夜,70-73%;v)10%aq.h2so4,thf,45℃,2h,73-78%;vi)胺(2.0当量),nabh(oac)3(2.0当量),acoh(2.0当量),dipea(2.0当量),1,2-二氯乙烷,rt,过夜。实施例通过下述实施例,进一步解释说明本发明。下面的实施例是非限制性的,并仅是本发明各方面的代表。本文公开的结构中实的和虚的楔形阐明相对立体化学,仅当特别说明或描绘时,描写绝对立体化学。通用方法利用与shimadzulc-2020质量检测器(电喷射离子化,esi)配对的shimadzulc-2030chplc系统,完成分析型高效液相-质谱(hplc-ms)。rp-hplc柱子是shimadzuc18(3um,50x4.6mm)。hplc方法1:50~100%乙腈水溶液(0.1%甲酸)作为流动相,流速1.0ml/min,以及5min运行时间。hplc方法2:10~100%乙腈水溶液(0.1%甲酸)作为流动相,流速1.0ml/min,以及10min运行时间。利用interchimpf4250系统完成制备型hplc纯化,并且通过254nm的uv吸收触发级分(fraction)收集。反相hplc柱是phenomenex00d-4454-u0-ax(gemini5umnx-c18,100x30mm)。prep-hplc方法1:5~85%乙腈水溶液作为流动相,流速35ml/min,以及20min运行时间。prep-hplc方法2:5~85%乙腈水溶液(0.05%三氟乙酸)作为流动相,流速35ml/min,以及20min运行时间。利用biotageisoleraone系统或interchimpf4250系统,完成正相快速柱色谱。硅胶柱购自teledyne(rediseprf)或biotage(snapultra)。流动相是不同比率的乙酸乙酯的己烷溶液或者甲醇的二氯甲烷溶液,并且254nm处的uv吸收触发级分收集。在biotageinitiator(最大400w,具有内部温度监控器)上,完成微波反应。所有原料和试剂是市售的,并直接使用。实施例1步骤1:化合物int-1-1c的合成将苯胺int-1-1a(1.52g,6.5mmol)、二唑酸int-1-1b(1.12g,7.9mmol)和pocl3(10ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃下,向所述残留物中加入h2o,并将混合物在0℃搅拌10min。将沉淀的粗氯喹int-1-1c过滤,用h2o洗涤,并减压干燥。没有进一步纯化,将粗产物int-1-1c用于下一步骤。步骤2:化合物int-1-1d的合成在室温下,向粗氯喹啉int-1-1c(2.33g,6.5mmol)和dipea(1.4ml,7.9mmol)的etoh(33ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.0ml,7.9mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩,并经柱色谱纯化(己烷/etoac梯度),以得到喹啉int-1-1d,作为深褐色固体物(1.0g,33%产率,通过两步)。lcms:(m+1)m/z=463,465.步骤3:化合物int-1-1e的合成在室温下,向化合物int-1-1d(0.85g,1.83mmol)、pd(oac)2(41mg,0.09mmol)和p(tbu)3·hbf4(32mg,0.11mmol)的thf(10ml)混悬液中,加入1mznbr2的thf(0.55ml)溶液。在室温下,向所述混合物中,历经30min,加入2miprmgcl的thf溶液(3.3ml,6.6mmol),并将得到的混合物在室温下搅拌过夜。将混合物倒入冰中,并用etoac(50ml)和1%aq.hcl(50ml)进行分配。将有机层用盐水(50ml)洗涤,na2so4干燥,并浓缩。残留物经柱色谱(1-10%etoac的ch2cl2溶液)纯化,然后经制备型-tlc纯化,以得到6-异丙基喹啉int-1-1e,作为黄色油状物(0.37g,47%产率)。lcms:(m+1)m/z=427.步骤4:化合物int-1-1f的合成在室温下,向int-1-1e(0.23g,0.55mmol)的thf(2.5ml)溶液中,加入10%aq.h2so4(5ml)。然后,将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(2×20ml)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-1-1f(黄色油状物)用于下一步反应。(210mg,定量产率)。lcms:(m+1)m/z=383.步骤5:化合物1的合成将int-1-1f(10.5mg,0.027mmol)、四氢-2h-吡喃-4-胺(0.054mmol)和dipea(10μl,0.054mmol)的1,2-二氯乙烷(1.5ml)混合物在室温下搅拌10min。向混合物中,加入nabh(oac)3(17.4mg,0.081mmol)和acoh(5μl,0.081mmol)。将得到的混合物在室温下搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。将残留物经制备型-tlc纯化(ch2cl2:meoh=96:4),得到化合物1(93%产率)。lcms:(m+1)m/z=468.实施例2–7将实施例1的方法用于生产化合物2–7,除了将步骤5的四氢-2h-吡喃-4-胺分别用(r)-四氢-2h-吡喃-3-胺代替,得到化合物2,lcms:(m+1)m/z=468;用(s)-四氢-2h-吡喃-3-胺代替,得到化合物3,lcms:(m+1)m/z=468);用(r)-四氢呋喃-3-胺代替,得到化合物4,lcms:(m+1)m/z=454);用(s)-四氢呋喃-3-胺代替,得到化合物5,lcms:(m+1)m/z=454);用环丙基甲胺代替,得到化合物6,lcms:(m+1)m/z=438);并用环丁基甲胺代替,得到化合物7,lcms:(m+1)m/z=452)。实施例8–10化合物8的合成利用类似于int-1-1d和int-1-1f合成所用的通用方法的条件,用两步,由int-11-42和1,4-二氧杂-8-氮杂螺[4,5]癸烷制备int-8-5。将int-8-5(11mg,0.029mmol)和环丙基甲胺(4.1mg,0.058mmol)混合物的1,2-二氯乙烷(0.2ml)溶液在rt搅拌10min。向所述混合物中,加入nabh(oac)3(12mg,0.058mmol)和acoh(3.3μl,0.058mmol)。将得到的混合物在rt搅拌过夜。将反应混合物减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物8,作为黄色固体物(8.8mg,0.020mmol)。lcms:(m+1)m/z=436,437.化合物9的合成将int-8-5(11mg,0.029mmol)、盐酸环丁基甲胺(7.0mg,0.058mmol)和dipea(10μl,0.058mmol)混合物的1,2-二氯乙烷(0.2ml)溶液在rt搅拌10min。向所述混合物中,加入nabh(oac)3(12mg,0.058mmol)和acoh(3.3μl,0.058mmol)。将得到的混合物在rt搅拌过夜。将反应混合物减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物9,作为黄色固体物(9.0mg,0.020mmol)。lcms:(m+1)m/z=450,451.化合物10的合成将int-8-5(11mg,0.029mmol)、(s)-四氢-2h-吡喃-3-胺盐酸盐(7.9mg,0.058mmol)和dipea(10μl,0.058mmol)混合物的1,2-二氯乙烷(0.2ml)溶液在rt搅拌10min。向所述混合物中,加入nabh(oac)3(12mg,0.058mmol)和acoh(3.3μl,0.058mmol)。将得到的混合物在rt搅拌过夜。将反应混合物减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物10,作为黄色固体物(9.7mg,0.021mmol)。lcms:(m+1)m/z=466,467.实施例11–1240的合成将int-11-1溴化物(1.0当量)、环丙基三氟硼酸钾(3.0当量)、k3po4(3.3当量)和pd(pph3)4(0.02当量)混合物的甲苯:h2o(3:1)溶液在100℃加热过夜。将混合物减压浓缩,并将产物经柱色谱纯化,利用己烷/ch2cl2(9:1)。得到产物,作为浅黄色固体物,产率39-41%。lcms:(m+1)m/z=194.42的合成将pocl3中的int-11-40(1.0当量)和int-11-41(1.0当量)的混合物在80℃加热1h。减压除去过量的pocl3。将残留物用冰淬灭,并将混合物搅拌15min。将浅棕色产物经过滤收集,并没有纯化,用于下一步骤(53-55%产率)。lcms:(m+1)m/z=318.化合物11的合成将乙醇中int-11-42(1.0当量)、int-11-44(2.0当量)和dipea(4.0当量)的混合物在130℃加热过夜。将混合物减压浓缩,并将产物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物11(产率62-64%),作为浅黄色油状物。lcms:(m+1)m/z=466.化合物12的合成将乙醇中int-11-42(1.0当量)、int-11-43(2.0当量)和dipea(2.0当量)的混合物在130℃加热过夜。将混合物减压浓缩,并将产物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物12(产率67-69%),作为浅黄色固体物。lcms:(m+1)m/z=466.实施例13–14根据化合物11和化合物12的方法,利用适当的中间体,得到化合物13和化合物14。得到作为浅黄色固体物的化合物13(产率69%,最后一步)。lcms:(m+1)m/z=452.得到作为浅黄色固体物的化合物14(产率73%,最后一步)。lcms:(m+1)m/z=452.实施例15–16int-15-2的合成向int-1-1d(227mg,0.49mmol)、环丁基三氟硼酸钾(159mg,0.98mmol)、cs2co3(479mg,1.47mmol)、catacxium(18mg,0.049mmol)和pd(oac)2(11mg,0.049mmol)的混合物中,加入甲苯(1.8ml)和h2o(0.16ml)。将反应混合物真空脱气,并填充氮气,然后在100℃加热过夜。将反应混合物冷却至rt,过滤(whatman注射式滤器,0.2μm孔径),干法装载至硅胶上,并经柱色谱纯化(己烷/etoac梯度),得到int-15-2,作为黄色固体物(86mg,产率39%)。lcms:(m+1)m/z=439.3的合成在室温下,向int-15-2(68mg,0.15mmol)的thf(0.5ml)溶液中,加入10%aq.h2so4(1.1ml)。然后,将混合物在45℃搅拌4h。冷却至rt后,将混合物用饱和na2co3水溶液中和,并用etoac(2×10ml)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-15-3用于下一步骤(56mg,定量产率)。lcms:(m+1)m/z=395.化合物15的合成将1,2-二氯乙烷(0.2ml)中int-15-3(10mg,0.025mmol)、(s)-四氢-2h-吡喃-3-胺盐酸盐(7mg,0.054mmol)和dipea(9μl,0.051mmol)的混合物在室温下搅拌10min。向混合物中,加入nabh(oac)3(11mg,0.051mmol)和acoh(3μl,0.051mmol)。将得到的混合物在室温下搅拌过夜。将得到的混合物减压浓缩。将残留物经制备型-tlc纯化(ch2cl2:meoh=95:5),得到化合物15(10.2mg,产率84%),作为黄色固体物。lcms:(m+1)m/z=480,481.化合物16的合成将1,2-二氯乙烷(0.2ml)中的int-15-3(10mg,0.025mmol)和(s)-四氢呋喃-3-胺(4.4mg,0.051mmol)的混合物在rt搅拌10min。向所述混合物中,加入nabh(oac)3(11mg,0.051mmol)和acoh(3μl,0.051mmol)。将得到的混合物在rt搅拌过夜。将反应混合物减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物16,作为黄色固体物(10mg,产率84%)。lcms:(m+1)m/z=466,467.实施例17向1-(6-环丁基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮(int-15-3,12mg,0.03mmol)和四氢-2h-吡喃-4-胺(6.2mg,0.061mmol)的1,2-二氯乙烷(0.2ml)溶液中,加入乙酸(3.7mg,0.061mmol),随后加入三乙酰氧基硼氢化钠(13mg,0.061mmol),并将反应混合物在25℃搅拌6h。将反应用甲醇淬灭,并用碳酸钠水溶液调至ph~10。然后,将混合物用乙酸乙酯萃取两次。将有机层合并,用盐水洗涤,硫酸镁干燥,并最后蒸发至干。将残留物经prep-hplc纯化,得到化合物17(8.0mg,55%产率),作为白色固体物。lcms:(m+1)m/z=480;保留时间:2.79min(方法1)。实施例18除了用(r)-四氢-2h-吡喃-3-胺盐酸盐(2.0eq)和n,n-二异丙基乙胺(2.0eq)替换四氢-2h-吡喃-4-胺,化合物18的制备与1-(6-环丁基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物17)相同。lcms:(m+1)m/z=480;保留时间:3.10min(方法1)。实施例19除了用(r)-四氢呋喃-3-胺盐酸盐(2.0eq)和n,n-二异丙基乙胺(2.0eq)替换四氢-2h-吡喃-4-胺,化合物19的制备与1-(6-环丁基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物17)相同。lcms:(m+1)m/z=466;保留时间:2.67min(方法1)。实施例20-29反应试剂和条件:i)dmf(2.5equiv.),pocl3(7.0equiv.),0to75℃,48h,36-39%;ii)nah2po4(5.0equiv.),naclo2(3.0equiv.),na2so3(4.0equiv.),ch3cn,94-98%;iii)a)15(1.0equiv.),socl2(3.0equiv.),ch2cl2,50℃,2h,b)乙酰胺肟;(1.2equiv.),dipea(1.2equiv.),二氧六环100℃,4h,48-54%;iv1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2equiv.),dipea(2.0equiv.),etoh,125℃,过夜,70-73%;v)16(1.0equiv.),et3b(2.0equiv.),cs2co3(2.0equiv.),pd(dppf)cl2.ch2cl2(1/20equiv.),thf,70℃,1.5h,27-29%;vi)1d%aq.h2so4,thf,45℃,2h,73-78%;vii)胺(2.0equiv.),nabh(oac)3(2.0equiv.),acoh(2.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷rt,过夜viii)cprmgbr(3.2equiv.),pd(oac)2(0.2equiv.),p(tbu)3·bf4(0.1equiv.),1mznbr2inthf(0.3equiv.),thf,rt,25-28%.int-20-14的合成在0℃,将dmf(2.5当量)缓慢添加至pocl3(7.0当量)中,将混合物在rt再搅拌30min,随后将酰胺int-20-13一次加入。将混合物在rt搅拌30min,然后在75℃搅拌48h。将混合物用冰水淬灭,并搅拌30min。收集固体int-20-14,并且不需进一步纯化而使用。lcms:(m+1)m/z=287,289.int-20-15的合成向醛int-20-14(1.0当量)在ch3cn的悬浮混合物中,缓慢加入nah2po4(5.0当量)的水溶液,随后加入naclo2(3.0当量)。将混合物在rt搅拌过夜。将混合物用na2so3淬灭,并在rt搅拌30min,随后用2mhcl酸化(ph1-2)。将产物用etoac萃取4次,用na2so4干燥,并减压浓缩。不需进一步纯化而使用粗int-20-15。lcms:(m-1)m/z=301,303.int-20-16的合成向ch2cl2中的酸int-20-15(1.0当量)和socl2(3.0当量)的混合物中,加入一滴dmf,并将反应物在50℃加热2h。将混合物减压浓缩。将粗品溶解于无水二氧六环中,并在0℃下,缓慢加入到乙酰胺肟(1.2当量)和dipea(1.2当量)的二氧六环溶液中。将混合物在rt搅拌30min,并在100℃搅拌4h。将混合物用水稀释,并收集固体物,并且不需进一步纯化而使用。lcms:(m+1)m/z=441,443.将前一步得到的产物(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2当量)和dipea(2.0当量)的etoh溶液在125℃加热过夜。冷却至室温后,将混合物减压浓缩,并经柱色谱(己烷/etoac)纯化,得到缩酮int-20-16,作为浅黄色固体物。lcms:(m+1)m/z=449,451.int-20-17的合成向int-20-16(1.0当量)、cs2co3(2.0当量)和pd(dppf)cl2·ch2cl2(1/20当量)的thf混悬液中,加入1met3b(2.0当量)的thf溶液,并将反应物在70℃加热1.5h。冷却至rt后,将粗品经硅藻土过滤,并经柱色谱纯化(己烷:etoac),得到int-20-17,作为浅黄色固体物。lcms:(m+1)m/z=399.int-20-18的合成将缩酮int-20-17(1.0当量)的thf和10%aq.h2so4溶液在45℃搅拌2h。冷却至rt后,将混合物用饱和naoh水溶液中和,并用etoac萃取。将合并的有机层用na2so4干燥,并减压浓缩,得到酮int-20-18,作为浅黄色固体物(产率90-95%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=355。化合物20-24的合成将1,2-二氯乙烷中的酮int-20-18(1.0当量)、胺(2.0当量)、dipea(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在rt搅拌过夜。将混合物经硅藻土过滤、减压浓缩,并经hplc纯化,得到所需产物。得到化合物20,作为浅黄色固体物(产率92-94%)。lcms:(m+1)m/z=440.得到化合物21,作为浅黄色固体物(产率78-80%)。lcms:(m+1)m/z=440.得到化合物22,作为浅黄色固体物(产率79-80%)。lcms:(m+1)m/z=440.得到化合物23,作为浅棕色固体物(产率82-83%)。lcms:(m+1)m/z=426.得到化合物24,作为浅黄色固体物(产率86-87%)。lcms:(m+1)m/z=426.int-20-19的合成在rt下,向化合物int-20-16(1.0当量)、pd(oac)2(0.2当量)和p(tbu)3·hbf4(0.1当量)在无水thf中的混悬液中,加入1mznbr2的thf溶液(0.3当量)。在rt下,向混合物中,历经30min,加入1mcprmgbr的thf(3.2当量)溶液,并将得到的混合物搅拌过夜。将混合物用冰/nh4cl溶液淬灭,并用etoac萃取。将有机层用盐水洗涤,na2so4干燥,并浓缩。因为观察到剩余溴化物int-20-16(~30%)和产物int-20-19的rf无差异,没有进一步纯化而使用粗产物。lcms:(m+1)m/z=411.int-20-20的合成将上述混合物的thf/10%aq.h2so4溶液在45℃搅拌2h。冷却至室温后,将混合物用饱和naoh水溶液中和,并用etoac萃取,na2so4干燥,并减压浓缩。产物经柱色谱利用己烷/etoac纯化,得到酮int-20-20,作为浅黄色固体物(产率20-25%)。lcms:(m+1)m/z=367.化合物25-29的合成将1,2-二氯乙烷中的酮int-20-20(1.0当量)、适当的胺(2.0当量)、dipea(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在rt搅拌过夜。将混合物经硅藻土过滤、减压浓缩,并经hplc纯化,得到标题化合物。得到化合物25,作为浅黄色固体物(产率87-88%)。lcms:(m+1)m/z=452.得到化合物26,作为浅黄色固体物(产率68-69%)。lcms:(m+1)m/z=452.得到化合物27,作为浅黄色固体物(产率72-73%)。lcms:(m+1)m/z=452.得到化合物28,作为浅黄色固体物(产率69-70%)。lcms:(m+1)m/z=438.得到化合物29,作为浅黄色固体物(产率75-76%)。lcms:(m+1)m/z=438.实施例30步骤1:向8-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷(int-1-1d,258mg,0.56mmol)的四氢呋喃(3ml)溶液中,加入硫酸溶液(10%aq.,3ml),并将反应物在45℃下搅拌3h。将混合物用碳酸钠溶液中和至ph10,并用乙酸乙酯萃取两次。将有机层合并,用盐水洗涤,用na2so4干燥,并减压浓缩,得到1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮(int-30-11f),作为棕色固体物(248mg),没有进一步纯化,将其用于下一步骤。lcms(esi):m/z419(m+h);保留时间:2.93min(方法1)。步骤2:除了用1-(6-环丁基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮代替1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮(int-30-11f),化合物30的制备与1-(6-环丁基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物17)相同。lcms(esi):m/z504(m+h);保留时间:2.34min(方法1)。实施例31化合物31的制备与实施例30的步骤2相同,其中用(r)-四氢-2h-吡喃-3-胺盐酸盐(2.0当量)和n,n-二异丙基乙胺(2.0当量)代替四氢-2h-吡喃-4-胺。lcms(esi):m/z504(m+h);保留时间:2.48min(方法1)。实施例32制备化合物32的方法与实施例30的步骤2相同,其中用(s)-四氢-2h-吡喃-3-胺盐酸盐(2.0当量)和n,n-二异丙基乙胺(2.0当量)代替四氢-2h-吡喃-4-胺。lcms(esi):m/z504(m+h);保留时间:2.45min(方法1)。实施例33-352-氨基-5-溴-3-氟苯甲酸int-33-14b的合成向2-氨基-3-氟苯甲酸int-33-14a(15.36g,99mmol)在ch2cl2(247.5ml)的混悬液中,加入n-溴代琥珀酰亚胺(17.62g,99mmol)。将混合物在室温搅拌过夜。将产物经真空过滤而收集,得到2-氨基-5-溴-3-氟苯甲酸int-33-14b,作为灰白色固体物(20.81g,产率90%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=232,234.1-(2-氨基-5-甲氧基-3-氟苯基)乙酮14f的合成将dmf(142ml)中的int-33-14b(13.38g,57.17mmol)、n,o-二甲基羟基胺盐酸盐(10g,102.9mmol)、dipea(19.9ml)、edci(13.15g,68.8mmol)和hobt(9.27g,68.6mmol)的混合物在室温搅拌4h。然后,将反应物用100mletoac稀释,并依次用1mnaoh、1mhcl和盐水洗涤,得到int-33-14c,作为棕色油状物(8.6g,产率85%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=277,279.在0℃,向int-33-14c(6.0g,22mmol)的ch2cl2(75ml)溶液中,加入tea(3.6ml,26mmol),随后滴加三氟乙酸酐(3.9ml,28mmol)。将反应物在室温搅拌过夜。然后,加入饱和nahco3水溶液,并分离有机相,并用水洗涤。将有机层用na2so4干燥,并浓缩至干,得到int-33-14d,作为黄色固体物(9.4g,80%产率),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=371,373.在-78℃,向int-33-14d(0.180g,0.554mmol)的thf(1ml)溶液中,加入3mmemgcl的thf(0.66ml,2.22mmol)溶液,并将反应物在室温搅拌2h。将反应物倒入冰中,用2mhcl酸化至ph2,并用etoac萃取。将合并的有机层用na2so4干燥,并浓缩至干,得到int-33-14e,作为黄色油状物,没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=328.向int-33-14e(0.138g,0.370mmol)的meoh(0.75ml)溶液中,加入2mnaoh溶液(0.75ml),将反应物在90℃加热1.5h。加入水,并将产物用etoac萃取,并浓缩至干。得到int-33-14f,作为黄色固体物(0.110g,产率92%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=232.2-氯喹啉int-33-14h的合成将2-(3-甲基-1,2,4-二唑-5-基)乙酸int-33-14g(72mg,0.50mmol)的pocl3(1.5ml)溶液在50℃搅拌5min后,加入酮int-33-14f(92mg,0.50mmol)。将混合物在110℃加热1h。真空除去过量的pocl3。向残留物中,加入饱和nahco3水溶液,并将产物用etoac萃取。将合并的有机层经na2so4干燥,并浓缩至干。粗产物经柱色谱(己烷/etoac)纯化,得到2-氯喹啉int-33-14h,作为黄色固体物(69mg,产率45%).lcms:(m+1)m/z=357.缩酮int-33-14i的合成向int-33-14h(56mg,0.182mmol)的etoh(0.4ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(29mg,0.200mmol)和dipea(38μl,0.218mmol)。将混合物置于110℃的微波辐射下,保持45min。将混合物减压浓缩,并经柱色谱(己烷/etoac)纯化,得到缩酮int-33-14i,作为黄色固体物(60mg,80%产率)。lcms:(m+1)m/z=464.8-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷int-33-14j的合成向微波波纹瓶中,加入int-33-14i(1.08g,2.89mmol)、pd2(dba)3(0.119g,0.130mmol)、tbuxphos(0.116g,0.275mmol)和kb(ome)4(1.51g,8.68mmol),并且加入dmf(3ml)之前,将气氛用氮气交换三次。将混合物加热至100℃,保持2h。冷却至室温后,将粗产物经硅藻土过滤,并经柱色谱(己烷/etoac)纯化,得到int-33-14j,作为黄色固体物(0.348g,37%产率)。lcms:(m+1)m/z=415.酮int-33-14k的合成向缩酮int-33-14j(60mg,0.145mmol)的thf(0.5ml)溶液中,加入10%h2so4水溶液(0.71ml)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取,经na2so4干燥,并浓缩,得到酮int-33-14k,作为黄色油状物(224mg,产率90%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=371.化合物33的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(10mg,0.027mmol)、3-(r)-氨基四氢吡喃hcl(5.6mg,0.040mmol)、dipea(4μl,0.030mmol)和acoh(2μl,0.054mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(11.5mg,0.0540mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物33,作为黄色固体物(4.3mg,35%产率)。lcms:(m+1)m/z=456.化合物34的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(20mg,0.054mmol)、3-(r)-氨基四氢呋喃(7.1mg,0.081mmol)和acoh(5μl,0.11mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(22.9mg,0.108mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物34,作为黄色固体物(10mg,42%产率)。lcms:(m+1)m/z=442.化合物35的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(10mg,0.027mmol)、3-氧杂环丁烷胺(3.0mg,0.040mmol)和acoh(2.0μl,0.054mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(11.5mg,0.0540mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物35,作为黄色固体物(2.5mg,22%产率)。lcms:(m+1)m/z=428.实施例36-39int-36-7的合成向微波波纹瓶中,加入int-33-14d(1.08g,2.89mmol)、pd2(dba)3(0.119g,0.130mmol)、tbuxphos(0.116g,0.275mmol)和kb(ome)4(1.51g,8.68mmol),并且将气氛用氮气交换三次,然后加入dmf(3ml)。将混合物加热至100℃,保持2h。冷却至室温后,将粗产物经硅藻土过滤,并经柱色谱(己烷/etoac)纯化,得到int-36-5,作为黄色固体物(0.348g,产率27.9%)。lcms:(m+1)m/z=325.在-78℃,向int-36-5(0.180g,0.554mmol)的thf(1ml)溶液中,加入3mmemgcl的thf溶液(0.66ml,2.22mmol),并将反应物在室温搅拌2h。将反应物倒入冰中,用2mhcl酸化至ph2,并用etoac萃取。将合并的有机层用na2so4干燥,并浓缩至干,得到int-36-6,作为黄色油状物,没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=280.向int-36-6(0.138g,0.370mmol)的meoh(0.75ml)溶液中,加入2mnaoh水溶液(0.75ml),将反应物在90℃加热1.5h。加入水,并将产物用etoac萃取,并真空浓缩至干。得到int-36-7,作为黄色固体物(0.110g,产率92%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=182.int-36-14h的合成将2-(3-甲基-1,2,4-二唑-5-基)乙酸int-33-14g(72mg,0.50mmol)的pocl3(1.5ml)溶液在50℃搅拌5min后,加入酮int-36-7(92mg,0.50mmol)。将混合物在110℃加热1h。真空除去过量的pocl3。向残留物中,加入饱和nahco3水溶液,并将产物用etoac萃取。将合并的有机层经na2so4干燥,并浓缩至干。粗产物经柱色谱(己烷/etoac)纯化,得到2-氯喹啉int-36-14h,作为黄色固体物(69mg,产率45%).lcms:(m+1)m/z=307,309.int-33-14j的合成向int-36-14h(56mg,0.18mmol)的etoh(0.4ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(29mg,0.200mmol)和dipea(38μl,0.22mmol)。将混合物置于110℃的微波辐射下,保持45min。将混合物减压浓缩,并经柱色谱(己烷/etoac)纯化,得到缩酮int-33-14j,作为黄色固体物(60mg,80%产率)。lcms:(m+1)m/z=415.int-33-14k的合成向缩酮int-33-14j(60mg,0.15mmol)的thf(0.5ml)溶液中,加入10%h2so4水溶液(0.71ml)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取,经na2so4干燥,并浓缩至干,得到酮int-33-14k,作为黄色油状物(224mg,产率90%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=371.化合物36的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(10mg,0.027mmol)、4-氨基四氢吡喃(4.1mg,0.040mmol)和acoh(2.0μl,0.054mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(11.5mg,0.0540mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物36,作为黄色固体物(2.5mg,产率20%)。lcms:(m+1)m/z=456.化合物37的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(10mg,0.027mmol)、3-(s)-氨基四氢吡喃hcl(4.1mg,0.030mmol)、dipea(4μl,0.030mmol)和acoh(2μl,0.054mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(11.5mg,0.0540mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物37,作为黄色固体物(2.3mg,产率18.7%)。lcms:(m+1)m/z=456.化合物38的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(10mg,0.027mmol)、3-(s)-氨基四氢呋喃(3.5mg,0.040mmol)和acoh(2μl,0.054mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(11.5mg,0.0540mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物38,作为黄色固体物(2.5mg,产率20%)。lcms:(m+1)m/z=442.化合物39的合成将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(10mg,0.027mmol)、环丙基甲胺(2.9mg,0.040mmol)和acoh(2μl,0.054mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(11.5mg,0.0540mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物39,作为黄色固体物(2.5mg,产率20%)。lcms:(m+1)m/z=426.实施例40–82化合物40的合成步骤1:将dmf(1.5ml)中的(3s,4r)-4-氨基-3-氟哌啶-1-甲酸叔丁酯(300mg,1.38mmol)、甲磺酸(s)-四氢呋喃-3-基酯(344mg,2.07mmol)和k2co3(286mg,2.07mmol)的混合物在72-85℃搅拌18h。溶剂蒸发后,将残留物经色谱(硅胶,0-10%meoh/dcm)纯化,得到(3s,4r)-3-氟-4-(((r)-四氢呋喃-3-基)氨基)哌啶-1-甲酸叔丁酯(129mg,32%产率),作为白色固体物。lcms(esi):m/z289(m+h);保留时间:1.47min(方法1)。步骤2:将上述产物(65mg,0.22mmol)的tfa(0.35ml,4.5mmol)和ch2cl2(1ml)溶液在25℃搅拌1h。将溶液用ch2cl2和meoh稀释,用nahco3(饱和水溶液)洗涤,mgso4干燥,并最后减压浓缩。没有进一步纯化而使用残留物。lcms(esi):m/z189(m+h).步骤3:本步骤类似于化合物134合成中的最后一步,溶剂选自二氧六环、乙醇、乙腈和/或dmf,并且温度是120℃~155℃。rp-hplc纯化后,得到化合物40(20mg,23%产率),作为黄色固体物。lcms(esi):m/z460(m+h);保留时间:2.00min(方法1)。根据上文对化合物40所公开的,利用适当的具有所需立体化学的氟-取代哌啶和适当的胺,得到化合物41-43。prep-tlc纯化后,得到化合物41(10.9mg,24%产率),作为白色固体物。lcms(esi):m/z460(m+h);保留时间:4.18min(方法2)。prep-tlc纯化后,得到化合物42(2.9mg,6.4%产率),作为白色固体物。lcms(esi):m/z460(m+h);保留时间:1.74min(方法1)。rp-hplc纯化后,得到化合物43(25mg,34%产率),作为黄色固体物。lcms(esi):m/z460(m+h);保留时间:1.88min(方法1)。如下文对化合物134-137所公开的,利用适当的具有所需立体化学的胺和上文所述的用于化合物36-39制备的中间体int-36-14h,得到化合物44-59。实施例55和58化合物55的合成rp-hplc纯化后,得到化合物55(1.3mg,产率7%),作为黄色固体物。lcms(esi):m/z474(m+h);保留时间:2.00min(方法1)。rp-hplc纯化后,得到化合物58(41mg,产率50%),作为灰白色固体物。lcms(esi):m/z444(m+h);保留时间:2.03min(方法1)。化合物60-61的合成根据对化合物40-59所述的方法,利用具有适当立体化学的胺int-60-3和实施例118中描述了其制备的中间体int-118-7,得到化合物60-61。化合物62-66的合成根据下文对化合物141所公开的由中间体int-62-1开始,以及根据对化合物35所用的合成方法,得到化合物62-66。化合物67-71的合成用上文对于化合物36-39公开的的方式,得到化合物67-71。化合物72-76的合成用上文对于化合物36-39公开的方式,由下文对于制备化合物156-158所述的中间体int-156-28开始,得到化合物72-76。化合物77-82的合成用上文对于化合物35公开的方式,得到化合物77-82。rp-hplc纯化后,得到为灰白色固体的化合物78。lcms(esi):m/z456(m+h);保留时间:4.00min(方法2)。rp-hplc纯化后,得到为灰白色固体的化合物80。lcms(esi):m/z470(m+h);保留时间:3.88min(方法2)。实施例83-84int-83-2的合成向int-33-14h(1009mg,2.830mmol)的etoh(0.4ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(449.8mg,3.141mmol)和dipea(0.59ml,3.4mmol)。将混合物置于120℃的微波辐射下,保持45min。将混合物减压浓缩,并经柱色谱(己烷/etoac)纯化,得到缩酮int-83-2,作为黄色油状物(1198mg,产率91.4%)。lcms:(m+1)m/z=463,465.int-83-3的合成向微波波纹瓶中,填充氯化烯丙基钯二聚物(2.0mg,0.0054mmol)、cs2co3(527.4mg,1.619mmol)、rockphos(7.6mg,0.015mmol)和int-83-2(500mg,1.07mmol)。将气氛用氮气交换三次,然后加入异丙醇(163ul,2.15mmol)和甲苯(2ml)。将混合物加热至90℃过夜。冷却至室温后,将粗产物经硅藻土过滤,并用etoac稀释。加入水,并分离各层。将水层用etoac萃取3次。将有机层合并,并用盐水洗涤,然后经na2so4干燥。真空除去溶剂,并将粗产物经反相色谱纯化(20-100%acn:h2o),得到int-83-3,作为黄色固体物(52g,产率15%)。lcms:(m+1)m/z=443.int-83-4的合成向缩酮int-83-3(60mg,0.14mmol)的thf(0.5ml)溶液中,加入10%h2so4水溶液(0.71ml)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取,经na2so4干燥,并浓缩至干,得到酮int-83-4,作为黄色油状物(224mg,产率90%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=399.化合物83的合成将1,2-二氯乙烷(0.1ml)中的酮int-83-4(10mg,0.025mmol)、3-(r)-氨基四氢吡喃hcl(5.2mg,0.038mmol)、dipea(4.0μl,0.03mmol)和acoh(2μl,0.050mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(10.6mg,0.0502mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物83,作为黄色固体物(1.83mg,产率15.1%)。lcms:(m+1)m/z=484.化合物84的合成将1,2-二氯乙烷(0.1ml)中的酮int-83-4(10mg,0.025mmol)、3-(r)-氨基四氢呋喃(3.3mg,0.037mmol)和acoh(2μl,0.050mmol)的混合物在室温搅拌15分钟后,加入nabh(oac)3(10.6mg,0.050mmol)。将混合物在室温搅拌5小时。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物84,作为黄色固体物(2.8mg,产率20%)。lcms:(m+1)m/z=470.实施例85int-85-40的合成向int-85-38(1.0当量)的ch2cl2溶液中,加入nbs(1.05当量),并将混合物在rt搅拌18h。将混合物浓缩,并用柱色谱利用ch2cl2纯化。lcms:(m+1)m/z=283,285.将二氧六环中的中间体醛(1.0当量)、int-85-39(2.0当量)和nh4oac(5.0当量)的混合物在90℃加热18h。将混合物浓缩,并将粗品用水(3x)稀释,并用己烷:etoac(9:1)(2x)萃取。将有机溶液减压浓缩,并且没有进一步纯化而使用产物。lcms:(m+1)m/z=332,334.int-85-41的合成将int-85-40(1.0当量)和pocl3的混合物在120℃加热18h。将混合物减压浓缩,用冰/水淬灭,并用nahco3碱化。将产物用ch2cl2(3x)萃取,将有机相用na2so4干燥并浓缩。没有进一步纯化而使用产物(int-85-41)。lcms:(m+1)m/z=350,352.int-85-42的合成将iproh中的int-85-41(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0当量)和dipea(2.0当量)的混合物在110℃加热18h。冷却至室温后,将混合物减压浓缩,并经柱色谱(etoac/己烷)纯化,得到相应的缩酮。lcms:(m+1)m/z=458,460.将iproh中的缩酮(1.0当量)、nh2oh.hcl(5.0当量)和na2co3(5.0当量)的混合物在120℃加热18h。将混合物冷却至rt、过滤并减压浓缩。没有进一步纯化即使用固体物。lcms:(m+1)m/z=491,493.将二氧六环中的酰氨肟(1.0当量)、ac2o(1.1当量)和dipea(1.1当量)的混合物在100℃加热18h。将混合物减压浓缩,并将产物经柱色谱(etoac/己烷)纯化,得到int-85-42。lcms:(m+1)m/z=515,517.int-85-43的合成将dmf中的int-85-42(1.0当量)、ki(1.0当量)、氰化锌(3.0当量)、pd(oac)2(0.2当量)、dppe(0.3当量)、na2co3(3.0当量)和tmeda(1.0当量)的混合物在130℃加热过夜。将混合物用etoac稀释,并用盐水(3x)洗涤。将有机相浓缩,并经制备型-tlc纯化产物。lcms:(m+1)m/z=462.将前述产物在thf/10%aq.h2so4中的混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和nahco3水溶液中和,并用etoac萃取产物(3x)。将有机相经na2so4干燥,并减压浓缩,得到酮int-85-43,没有进一步纯化而使用。lcms:(m+1)m/z=418.化合物85的合成将1,2-二氯乙烷中的酮int-85-43(1.0当量)、4-氨基四氢吡喃(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在室温搅拌过夜。将混合物经硅藻土过滤,并减压浓缩,并经hplc纯化,得到化合物85,作为黄色固体物(产率84-86%)。lcms:(m+1)m/z=503.实施例86根据下文对化合物88公开的方法,由酮int-88-24和适当的胺起始,得到化合物86,作为浅黄色固体物(产率49-51%)。lcms:(m+1)m/z=470.实施例87int-87-2的合成向2-氨基-3-氯苯甲酸int-87-1(4.57g,26.6mmol)在ch2cl2(100ml)的混悬液中,加入n-溴代琥珀酰亚胺(4.74g,26.6mmol)。将混合物在室温搅拌2.5h。将产物经过滤,用ch2cl2洗涤,并减压干燥,得到int-87-2,作为灰白色固体物(5.67g,产率85%)。lcms:(m-1)m/z=248,250.int-87-3的合成在0℃,向int-87-2(5.41g,21.6mmol)的thf(50ml)溶液中,滴加1mbh3.thf络合物在thf中的溶液(108ml,108mmol)。然后,将混合物在室温搅拌18h。通过在0℃缓慢加入meoh而淬灭反应。减压除去挥发性物质后,将残留物在etoac和盐水间进行分配。将有机层用na2so4干燥并浓缩。没有进一步纯化,而将产物int-87-3(作为粉色固体物)用于下一步反应(定量产率)。int-87-4的合成将ch2cl2(100ml)中的int-87-3(5.10g,21.6mmol)和活化mno2(11.2g,129.4mmol)的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液浓缩。将残留物经短硅胶柱纯化,得到int-87-4,作为黄色固体物(5.06g,定量产率)。int-87-6的合成将醛int-87-4(1.52g,6.48mmol)、噁二唑酯int-87-5(1.10g、6.48mmol)和p-tsa(0.15g,10wt%)的混合物在150℃加热1h。冷却至室温后,加入ch2cl2:meoh(9:1,v/v,20ml)和己烷:etoac(8:2,v/v,20ml),并将得到的混合物剧烈搅拌。目标产物在溶剂减压蒸发期间,从溶液中析出。将固体物过滤,用h2o洗涤,并减压干燥。没有进一步纯化,而将产物6(作为棕色固体物)用于下一步反应(338mg,定量产率15%)。lcms:(m+1)m/z=340,342.int-87-7的合成将int-87-6(0.50g,1.47mmol)和pocl3(5ml)的混合物在110℃加热1h。冷却至室温后,减压除去过量的pocl3。在0℃,向残留物中加入h2o。将混合物在0℃搅拌10min。将沉淀的固体物过滤,用h2o洗涤,并减压干燥,得到int-87-7,作为浅棕色固体,没有进一步纯化而将其用于下一步骤(447mg,产率85%)。int-87-8的合成将etoh(7.6ml)中的int-87-7(272mg,0.76mmol)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(0.20ml,1.52mmol)和dipea(0.26ml,1.52mmol)的混合物在120℃加热过夜。冷却至室温后,将混合物浓缩,并经柱色谱(己烷/etoac梯度)纯化,得到int-87-8,作为黄色油状物(287mg,产率82%).lcms:(m+1)m/z=465,467.int-87-9的合成在室温下,向int-87-8(287mg,0.62mmol)、k2co3(170mg,1.24mmol)和pd(pph3)4(71mg,0.062mmol)在thf(12ml)中的混合物中,加入1met3b的thf溶液(1.3ml,1.24mmol)。然后,将混合物在70℃加热过夜。冷却至室温后,将混合物经硅藻土过滤。将滤液浓缩,并经柱色谱(己烷/etoac梯度)纯化,得到int-87-9,作为黄色油状物(70mg,产率27%).lcms:(m+1)m/z=415.int-87-10的合成向int-87-9(70mg,0.17mmol)的thf(1ml)溶液中,加入10%h2so4水溶液(2ml,v/v)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(×2)。将合并的有机萃取物用na2so4干燥,并浓缩至干。没有进一步纯化,将黄色固体产物int-87-10用于下一步反应(62mg,定量产率)。lcms:(m+1)m/z=371.化合物87的合成将1,2-二氯乙烷(1.0ml)中的酮int-87-10(10.9mg,0.029mmol)、四氢-2h-吡喃-4-胺(6.0mg,0.058mmol)和dipea(10μl,0.058mmol)的混合物在室温搅拌10min。向所述混合物中,加入nabh(oac)3(18.4mg,0.087mmol)和acoh(5μl,0.087mmol)。将得到的混合物在室温搅拌18h。经硅藻土过滤,将滤液浓缩。将残留物经制备型-tlc(etoac:iproh=9:1)纯化,得到化合物87,作为黄色油状物(12.0mg,产率91%)。lcms:(m+1)m/z=456.实施例88反应试剂和条件:i)dmf(2.5equiv.),pocl3(7.0equiv.),0to75℃,48h,36-39%;ii)nah2po4(5.0equiv.),naclo2(3.0equiv.),na2so3(4.0equiv.),ch3cn,94-98%;iii)a)int-20-15(1.0equiv.),socl2(3.0equiv.),ch2cl2,50℃,2h,b)乙酰胺肟(1.2equiv.),dipea(1.2equiv.),二氧六环,100℃,4h,48-54%;iv1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2equiv.),dipea(2.0equiv.),etoh,125℃,过夜,70-73%;v)int-20-16(1.0equiv.),et3b(2.0equiv.),cs2co3(2.0equiv.),pd(dppf)cl2.ch2cl2(1/20equiv.),thf,70℃,1.5h,27-29%;vi)10%aq.h2so4,thf,45℃,2h,73-78%;vii)胺(2.0equiv.),nabh(oac)3(2.0equiv.),acoh(2.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷,rt,过夜;viii)cprmgbr(3.2equiv.),pd(oac)2(0.2equiv,),p(tbu)3·hbf4(0.1equiv.),1mznbr2inthf(0.3equiv.),thf,rt,25-28%.int-88-15的合成将酸int-88-14(1.0当量)和nbs(1.0当量)在ch2cl2中的混悬液在室温搅拌过夜。经过滤收集产物,得到酸int-88-15,作为灰白色固体物(产率84-86%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=247,249.int-88-16的合成将dmf中的int-88-15(1.0当量)、n,o-二甲基羟基胺盐酸盐(1.8当量)、dipea(2.0当量)、edci(1.2当量)和hobt(1.2当量)的混合物在室温搅拌4h。将反应物用etoac稀释,并依次用饱和nahco3水溶液和盐水洗涤,并真空浓缩,得到int-88-16,作为棕色油状物(产率84-86%)。没有进一步纯化,将产物用于下一步骤。lcms:(m-1)m/z=292,294.int-88-17的合成在0℃,向int-88-16(1.0当量)和tea(1.2当量)的ch2cl2溶液中,滴加三氟乙酸酐(1.3当量)。将反应物在室温搅拌过夜。将混合物用饱和nahco3水溶液和盐水洗涤(2x)。有机层经na2so4干燥,并浓缩,得到int-88-17,作为浅黄色固体物(产率90-92%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=386,388.int-88-18的合成向int-88-17(1.0当量)、cs2co3(3.0当量)和pd(dppf)cl2(0.02当量)在thf中的混悬液中,加入1met3b的thf溶液(3.0当量),并将反应物在70℃加热1.5h。冷却至室温后,将粗品经硅藻土过滤,并经柱色谱(己烷:etoac)纯化,得到int-88-18,作为黄色固体物(产率45-47%).lcms:(m+1)m/z=339.int-88-19的合成在0℃,向int-88-18(1.0当量)的thf溶液中,加入1.4mmemgbr的thf:甲苯溶液(5.0当量),并将反应物在室温搅拌2.5h。用冰淬灭反应,用2mhcl酸化至ph2,并用etoac萃取(2x)。将合并的有机层用na2so4干燥,并浓缩,得到int-88-19,作为黄色固体物(产率85-87%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=294.int-88-20的合成向int-88-19(1.0当量)的meoh溶液中,加入2mnaoh水溶液(1.7当量),并将反应物在90℃加热3h。将混合物用水稀释,并经过滤收集固体物,得到int-88-20,作为黄色固体物(产率80-82%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=198.int-88-21的合成将酮int-88-20(1.0当量)、int-88-8(2.0当量)和p-tsoh(cat.)的混合物在150℃搅拌2h。冷却至室温后,将反应混合物依次用etoac:己烷(8:2,v/v)(2x)和水洗涤,得到喹诺酮int-88-21,作为黄色固体物(产率19-21%).lcms:(m+1)m/z=304.int-88-22的合成将int-88-21的pocl3混悬液在110℃搅拌2h。减压除去过量的pocl3。将粗品用饱和nahco3水溶液淬灭,并将产物用etoac萃取(3x)。将合并的有机层经na2so4干燥,并浓缩。粗产物经柱色谱(己烷/etoac)纯化,得到2-氯喹啉int-88-22,作为浅黄色固体物(产率97-98%).lcms:(m+1)m/z=322,324.int-88-23的合成将etoh中的int-88-22(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0当量)和dipea(2.0当量)的混悬液在110℃加热过夜。冷却至室温后,将混合物减压浓缩,并经柱色谱(己烷/etoac)纯化,得到缩酮int-88-23,作为浅黄色固体物(产率73-75%).lcms:(m+1)m/z=429.int-88-24的合成向缩酮int-88-23的thf溶液中,加入10%h2so4水溶液,并将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(3×),用na2so4干燥,并浓缩,得到酮int-88-24,作为黄色固体(产率83-85%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=385.化合物88的合成将1,2-二氯乙烷中的酮int-88-24(1.0当量)、4-氨基四氢吡喃(1.5当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在室温搅拌过夜。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物88,作为黄色固体物(产率89-90%)。lcms:(m+1)m/z=470.实施例89根据下文对化合物91公开的方法,以96%产率(最后一步)得到化合物89,作为浅黄色固体物。lcms:(m+1)m/z=447.实施例90int-90-11的合成向2-氨基-5-溴苯甲酸int-90-8(10.80g,50.0mmol)的ch2cl2(200ml)混悬液中,加入n-碘代琥珀酰亚胺(11.25g,50.0mmol)。将混合物在室温搅拌4h。将产物过滤,用ch2cl2洗涤,并减压干燥,得到碘代苯甲酸int-90-9,作为浅棕色固体物。在0℃,向int-90-9(13.51g,39.5mmol)的thf(100ml)溶液中,滴加bh3.thf络合物溶液(1mthf溶液,200ml,200mmol)。然后,将混合物在室温搅拌过夜。在0℃,通过缓慢加入meoh而淬灭反应。将溶剂除去至干燥后,将粗醇int-90-10溶解于ch2cl2(200ml)中,并加入活化的mno2(20.60g,237mmol)。将混合物在室温搅拌过夜。将粗产物用短硅胶通路纯化,得到醛int-90-11,作为黄色固体物(10.3g,历经三步的产率63%)。lcms:(m-1)m/z=324,326.int-90-14的合成将醛int-90-11(4.50g,13.8mmol)、二唑酸int-90-12(2.36g,16.6mmol)和pocl3(14ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃,向所述残留物中加入h2o。将混合物在0℃搅拌10min。将粗氯化物int-90-13过滤,用h2o洗涤,并减压干燥。室温下,向int-90-13在etoh(70ml)中的混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(3.5ml,27.6mmol)和dipea(4.8ml,27.6mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩,并经柱色谱(己烷/etoac梯度)纯化,得到喹啉int-90-14,作为黄色油状物(2.61g,34%产率,通过两步).lcms:(m+1)m/z=557,559.int-90-15的合成将int-90-14(2.35g,4.22mmol)、zn(cn)2(0.50g,4.22mmol)、zn粉(30mg,0.42mmol)和pd(pph3)4(0.50g,0.42mmol)在thf/dmf(60ml,1:1(v/v))中的混合物于80℃加热过夜。冷却至室温后,将混合物经硅藻土过滤,并浓缩滤液。将残留物经柱色谱(己烷/etoac)纯化,得到氰基喹啉int-90-15,作为黄色油状物(0.86g,45%产率)。lcms:(m+1)m/z=456,458.int-90-16的合成室温下,向int-90-15(0.86g,1.88mmol)、k2co3(0.52g,3.76mmol)和pd(pph3)4(0.22g,0.19mmol)在dmf(20ml)的混合物中,加入1met3b的thf溶液(3.8ml,3.80mmol)。然后,将混合物在70℃加热过夜。冷却至室温后,将混合物在盐水(30ml)和etoac(30ml)之间进行分配。用etoac(30ml)萃取水层,并将合并的有机层用na2so4干燥,并浓缩。将残留物经柱色谱纯化(己烷/etoac梯度),得到乙基喹啉16,作为黄色油状物(0.23g,30%产率)。lcms:(m+1)m/z=406.int-90-17的合成向int-90-6(0.14g,0.35mmol)的thf(1ml)溶液中,加入10%aq.h2so4(5ml,v/v)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(2×20ml)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-90-7用于下一步反应(116mg,92%产率)。lcms:(m+1)m/z=362.化合物90的合成将1,2-二氯乙烷(1.5ml)中的酮int-90-17(14.4mg,0.04mmol)、(s)-3-氨基四氢吡喃盐酸盐(11.0mg,0.08mmol)和dipea(14μl,0.08mmol)的混合物在rt搅拌10min。向所述混合物中,加入nabh(oac)3(25.4mg,0.12mmol)和acoh(7μl,0.12mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物90,作为黄色油状物(15.6mg,产率88%)。lcms:(m+1)m/z=447.实施例91反应试剂和条件i)int-91-18(1.0equiv.),pylcl(1.0equiv.),meoh,回流18h,69%;ii)int-91-19(1.0equiv.),int-91-12(1.2equiv.),pocl3,110℃,1h;iii)1,4-二氧杂-8-氮杂螺「4,5]癸烷(2.0equiv.),dipea(2.0equiv.),etoh,120℃,18h,30%(两步);iv)zn(cn)2(1.0equiv.),zn(0.1equiv.),pd(pph3)4(0.1equiv.),thf/dmf,80℃,18h,86%;v)et3b(2.0equiv.),k2co3(2.0equiv.),pd(pph3)4(0.1equiv.),dmf,70℃,57%;vi)10%aq.h2so4,thf,45℃,2h,89%;vii)胺(2.0equiv.),nabh(oac)3(3.0equiv.),acoh(3.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷,rt,18h,93%.int-91-19的合成将meoh(54ml)中的2-氨基-5-溴苯乙酮int-91-18(4.6g,21.5mmol)和碘氯化吡啶鎓(pyridiniumiodochloride)(5.2g,21.5mmol)的混合物回流过夜。冷却至室温后,将混合物浓缩。残留物经柱色谱(己烷/etoac梯度)纯化,得到碘苯乙酮int-91-19,作为黄色固体物(5.04g,产率69%).lcms:(m+1)m/z=340,342.int-91-21的合成将酮int-91-19(5.02g,14.8mmol)、二唑酸int-91-12(2.52g,17.7mmol)和pocl3(15ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃,向所述残留物中加入h2o。将混合物在0℃搅拌10min。将粗氯化物int-91-20过滤,用h2o洗涤,并减压干燥。室温下,向int-91-20在etoh(74ml)中的混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(3.8ml,29.6mmol)和dipea(5.2ml,29.6mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩,并经柱色谱(己烷/etoac梯度)纯化,得到喹啉int-91-21,作为黄色油状物(2.54g,30%产率,通过两步).lcms:(m+1)m/z=571,573.int-91-22的合成将int-91-12(1.28g,2.24mmol)、zn(cn)2(0.26g,2.24mmol)、zn粉(15mg,0.22mmol)和pd(pph3)4(0.26g,0.22mmol)在thf/dmf(22ml,1:1(v/v))中的混合物于80℃加热过夜。冷却至室温后,将混合物经硅藻土过滤,并浓缩滤液。将残留物经柱色谱(己烷/etoac)纯化,得到氰基喹啉int-91-22,作为浅绿色固体物(0.90g,产率86%)。lcms:(m+1)m/z=470,472.int-91-23的合成室温下,向int-91-22(0.90g,1.91mmol)、k2co3(0.53g,3.82mmol)和pd(pph3)4(0.22g,0.19mmol)在dmf(20ml)的混合物中,加入1met3b的thf溶液(3.8ml,3.80mmol)。然后,将混合物在70℃加热过夜。冷却至室温后,将混合物在盐水(30ml)和etoac(30ml)之间进行分配。用etoac(30ml)萃取水层,并将合并的有机层用na2so4干燥,并浓缩。将残留物经柱色谱纯化(己烷/etoac梯度),得到乙基喹啉int-91-23,作为黄色油状物(0.45g,75%产率)。lcms:(m+1)m/z=420.int-91-24的合成向int-91-23(0.45g,1.08mmol)的thf(4ml)溶液中,加入10%aq.h2so4(12ml,v/v)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(2×30ml)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将为橙色固体的酮int-91-24用于下一步反应(0.36g,产率89%)。lcms:(m+1)m/z=376.化合物91的合成将1,2-二氯乙烷(1.5ml)中的酮int-91-24(15.1mg,0.04mmol)、(r)-3-氨基四氢吡喃盐酸盐(11.0mg,0.08mmol)和dipea(14μl,0.08mmol)的混合物在室温搅拌10min。向所述混合物中,加入nabh(oac)3(25.4mg,0.12mmol)和acoh(7μl,0.12mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。将残留物经制备型-tlc(etoac:iproh=9:1)纯化,得到化合物91,作为黄色油状物(17.2mg,产率93%)。lcms:(m+1)m/z=461.实施例92-93反应试剂和条件:i)int-92-1(1.0equiv.),int-142-9(1.5equiv.),nah(1.0equiv.),dmf,120℃,4h;ii)pocl3,110℃,1.5h,19%,两步;iii)int-92-4(1.0equiv.),1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0equiv.),pd(pph3)4(0.1equiv.),1,4-二氧六环,80℃,3h,11%forint-92-5(and48%forint-92-6);iv)5(1.0equiv.),et3b(1.2equiv.),k2co3(2.0equiv.),pd(pph3)4(0.1equiv.),thf/dmf,70℃,过夜67%;v)10%aq.h2so4,thf,45℃,2h,80%;vi)int-92-8(1.0equiv.),胺(2.0equiv.),nabh(oac)3(3.0equiv.),acoh(3.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷,rt,过夜,62%;vii)7(1.0equiv.),kcn(2.0equiv.),dmso,110℃,过夜,30%;viii)10%aq.h2so4,thf,45℃,1.5h,定量产率;ix)int-92-11(1.0equiv.),胺(2.0equiv.),nabh(oac)3(3.0equiv.),acoh(3.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷rt,18h,61%.int-92-3的合成在0℃,向nah(60%在油中,0.83g,20.7mmol)的dmf(60ml)混悬液中,滴加噁二唑酯int-92-2(5.30g,31.0mmol)。将混合物在0℃搅拌10min,随后加入5-溴靛红酸酐int-92-1(5.00g,20.7mmol)。然后,将混合物在120℃加热4h。冷却至室温后,通过加入h2o淬灭反应,并用浓hcl酸化(ph=4~5,经ph试纸)。将混合物用ch2cl2(×3)萃取,并将合并的有机萃取物用na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-92-3(暗褐色油状物)用于下一步反应。int-92-4的合成将int-92-3(6.67g,20.7mmol)和pocl3(5ml)的混合物在110℃搅拌1.5h。冷却至室温后,减压除去过量的pocl3。在0℃,向所述残留物中加入h2o。将混合物在ch2cl2和饱和nahco3水溶液之间进行分配。分离水层,并用ch2cl2(×2)萃取。将合并的有机层用na2so4干燥,并浓缩,残留物经柱色谱(己烷/etoac梯度)纯化,得到int-92-4,作为黄色固体物(1.41g,19%产率,两步)。int-92-5的合成将1,4-二氧六环(80ml)中的int-92-4(4.29g,11.95mmol)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(3.0ml,23.90mmol)和pd(pph3)4(1.38g,1.12mmol)的混合物在80℃加热3h。冷却至室温后,将混合物经硅藻土过滤。将滤液浓缩,并经柱色谱纯化(己烷/etoac梯度),得到int-92-5,作为浅黄色固体物(0.59g,11%产率),以及4-氨基化的int-92-6,作为浅黄色晶体(2.67g,48%产率)。经x-射线晶体学确定int-92-6的结构。lcms:(m+1)m/z=465,467.int-92-7的合成室温下,向thf/dmf(1∶1,v/v,12ml)中的int-92-5(590mg,1.27mmol)、k2co3(350mg,2.54mmol)和pd(pph3)4(146mg,0.127mmol)混合物中,加入1met3b的thf溶液(1.52ml,1.52mmol)。然后,将混合物在70℃加热过夜。冷却至室温后,将混合物经硅藻土过滤。将滤液浓缩,并经柱色谱纯化(己烷/etoac梯度),得到int-92-7,作为黄色油状物(352mg,产率67%)。lcms:(m+1)m/z=415.int-92-8的合成向int-92-7(125mg,0.30mmol)的thf(3ml)溶液中,加入10%aq.h2so4(5ml,v/v)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(×2)萃取。将合并的有机层用na2so4干燥,并浓缩。将残留物经柱色谱(己烷/etoac梯度)纯化,得到int-92-8,作为黄色油状物(88.7mg,产率80%)。lcms:(m+1)m/z=371.化合物92的合成将1,2-二氯乙烷(1.5ml)中的酮int-92-8(10.0mg,0.027mmol)、4-氨基四氢吡喃(5.5mg,0.054mmol)和dipea(10μl,0.054mmol)的混合物在室温搅拌10min。向所述混合物中,加入nabh(oac)3(17.0mg,0.081mmol)和acoh(5μl,0.081mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=93:7)纯化,得到化合物92,作为黄色油状物(7.6mg,产率62%)。lcms:(m+1)m/z=456.int-92-10的合成将dmso(10ml)中的int-92-7(334mg,0.805mmol)和kcn(105mg,1.61mmol)的混合物在110℃加热过夜。冷却至室温后,将混合物在etoac和盐水之间进行分配。分离水层,并用etoac(×3)萃取。将合并的有机层用na2so4干燥,并浓缩。残留物经柱色谱(己烷/etoac梯度)纯化,随后经制备型-tlc(己烷:etoac=6:1)纯化,得到int-92-10,作为黄色固体物(97mg,30%产率)。lcms:(m+1)m/z=406.int-92-11的合成向int-92-10(23mg,0.057mmol)的thf(1ml)溶液中,加入10%aq.h2so4(1ml,v/v)。将混合物在45℃搅拌1.5h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(×2)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-92-11(黄色油状物)用于下一步反应(20.5mg,定量产率)。lcms:(m+1)m/z=362.化合物93的合成将1,2-二氯乙烷(1.5ml)中的酮int-92-11(10.2mg,0.028mmol)、(r)-3-氨基四氢吡喃(7.7mg,0.056mmol)和dipea(10μl,0.056mmol)的混合物在室温搅拌10min。向所述混合物中,加入nabh(oac)3(17.0mg,0.084mmol)和acoh(5μl,0.084mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物93,作为黄色油状物(7.7mg,产率61%)。lcms:(m+1)m/z=447.实施例94-96int-94-2的合成将int-33-14h(1.753g,4.92mmol)和三丁基-(1-乙氧乙烯基)锡(2.33ml,6.89mmol)的甲苯(15ml)溶液用氮气脱气。加入双(三苯基膦)二氯化钯(ii)(172.7mg,0.245mmol,5mol%),并将反应混合物在80℃加热过夜。将反应混合物冷却至rt,并在硅藻土垫上过滤。将滤液蒸发,并将残留物经硅胶柱色谱纯化(己烷:etoac=90:1),得到int-94-2,作为白色固体物(537mg,31%产率)。lcms:(m+1)m/z=348,350.int-94-3的合成向1,4-二氧六环(3ml)的int-94-2(537mg,1.54mmol)混合物中,加入2m盐酸(3ml)。将反应混合物在rt搅拌30min。减压除去1,4-二氧六环。经真空过滤收集产物,依次用水和己烷清洗,得到int-94-3,作为灰白色固体物(480mg,97%产率)。lcms:(m+1)m/z=320,322.int-94-4的合成在0℃,向二氯甲烷(1.5ml)中的int-94-3(100mg,0.31mmol)混合物中,加入二乙基胺三氟化硫(0.25ml,1.88mmol)。将反应混合物在rt搅拌过夜。在0℃,加入另外的二乙基胺三氟化硫(0.21ml,1.58mmol),并将反应物在rt再搅拌48h。将反应混合物缓慢倒入饱和nahco3水溶液中。分离有机层,并将水层用二氯甲烷(3x20ml)萃取。将合并的有机层用na2so4干燥,并减压蒸发溶剂。将残留物经制备型-tlc纯化(己烷:etoac=80:20),得到int-94-4,作为白色固体物(62.4mg,产率59%).lcms:(m+1)m/z=342,344.int-94-5的合成向int-94-4(40mg,0.12mmol)的etoh(0.5ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(0.030ml,0.24mmol)和dipea(0.041ml,0.24mmol)。将反应混合物在130℃微波下加热40min。将混合物浓缩,并经制备型-tlc(己烷:etoac=80:20)纯化,得到int-94-5,作为黄色固体物(43mg,89%产率)。lcms:(m+1)m/z=449.int-94-6的合成在rt向int-94-5(43mg,0.096mmol)的thf(0.4ml)溶液中,加入10%aq.h2so4(0.8ml)。然后,将混合物在45℃搅拌4h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(2×10ml)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-94-6(38mg,98%;黄色油状物)用于下一步骤。lcms:(m+1)m/z=405.化合物94的合成将1,2-二氯乙烷(0.2ml)中的int-94-6(11mg,0.027mmol)、四氢-2h-吡喃-4-胺(5.5mg,0.054mmol)的混合物在rt搅拌10min。向所述混合物中,加入nabh(oac)3(11.4mg,0.054mmol)和acoh(3μl,0.054mmol)。将得到的混合物在rt搅拌过夜。将反应混合物直接经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物94,作为灰白色固体物(6.0mg,产率45%)。lcms:(m+1)m/z=490.化合物95的合成将1,2-二氯乙烷(0.2ml)中的int-94-6(11mg,0.027mmol)、(r)-四氢-2h-吡喃-3-胺盐酸盐(7.4mg,0.054mmol)和dipea(9.4μl,0.054mmol)的混合物在rt搅拌10min。向所述混合物中,加入nabh(oac)3(11.4mg,0.054mmol)和acoh(3μl,0.054mmol)。将得到的混合物在rt搅拌过夜。将反应混合物直接经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物95,作为白色固体物(6.0mg,产率45%)。lcms:(m+1)m/z=490.化合物96的合成将1,2-二氯乙烷(0.2ml)中的int-94-6(11mg,0.027mmol)和(r)-四氢呋喃-3-胺(4.7mg,0.054mmol)的混合物在rt搅拌10min。向所述混合物中,加入nabh(oac)3(11.4mg,0.054mmol)和acoh(3μl,0.054mmol)。将得到的混合物在rt搅拌过夜。将反应混合物直接经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物96,作为灰白色固体物(2.0mg,产率16%)。lcms:(m+1)m/z=476.实施例97-98int-97-3的合成将1,2-二氯乙烷(20ml)中的int-97-1(1.37g,6.89mmol)和四氢-2h-吡喃-4-胺int-97-2(1.395g,13.79mmol)的混合物在rt搅拌10min。向所述混合物中,加入nabh(oac)3(2.92g,3.79mmol)和acoh(0.78ml,13.79mmol)。将反应混合物在rt搅拌过夜。将溶剂减压蒸发,并将残留物在饱和na2co3水溶液和etoac之间进行分配。分离有机相,并将水层用etoac(2x40ml)萃取。将合并的有机相经na2so4干燥,并减压蒸发。将产物经柱色谱(ch2cl2:meoh=97:3)纯化,得到int-97-3,作为黄色油状物(1.716g,产率88%)。lcms:(m+1)m/z=285.int-97-4的合成向int-97-3(1.716g,6.03mmol)的ch2cl2(10ml)溶液中,滴加三氟乙酸(9.2ml,120.67mmol)。将反应混合物在rt搅拌1h,然后减压蒸发溶剂和过量的三氟乙酸。将残留物在2mnaoh溶液和etoac之间进行分配。分离有机相,并将水相用etoac(3x20ml)反萃取。将合并的有机相用水(2x5ml)洗涤,na2so4干燥,并减压蒸发,得到int-97-4,作为黄色油状物(1.1g,定量产率),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=185.int-97-5的合成与上文对化合物94-96所述的中间体int-94-3的合成相同。int-97-6的合成将二氯(p-伞花烃)钌(ii)二聚体(0.38mg,0.6μmol)和(1r,2r)-(-)-n-p-甲苯磺酰基-1,2-二苯基乙二胺(0.55mg,1.5μmol)悬浮于水(0.25ml)中。将混合物用氮气脱气,然后在氮气气氛下,于70℃加热90min。将得到的混合物冷却至rt。加入酮int-97-5(40mg,0.125mg)、甲酸钠(42.5mg,0.625mmol)和无水thf(0.12ml),并将反应物用氮气脱气。将反应混合物在40℃搅拌2.5h。将反应混合物用etoac稀释,并用盐水洗涤。将水相用etoac萃取。合并的有机层经na2so4干燥,并减压蒸发。将残留物经制备型-tlc(己烷:etoac=70:30)纯化,得到标题化合物,作为白色固体物(33.0mg,82%产率,ee未测定).lcms:(m+1)m/z=322,324.int-97-7的合成–78℃下,将n-(三甲基硅烷基)吗啉(60μl,0.336mmol)滴加到dast(43μl,0.328mmol)的干燥ch2cl2(0.2ml)溶液中。将得到的溶液在rt搅拌2.5h。将反应混合物在–78℃冷却,并滴加(r)-1-(2-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-6-基)乙-1-醇int-97-6(33mg,0.10mmol)的干燥ch2cl2(0.4ml)溶液。将反应混合物在rt搅拌过夜。然后,将反应混合物缓慢倒入10ml饱和nahco3溶液中。分离有机层,并将水相再用ch2cl2(3x10ml)萃取。将合并的有机相经na2so4干燥,并减压蒸发。将残留物经制备型-tlc(己烷:etoac=80:20)纯化,得到目标化合物,作为黄色油状物(14.6mg,45%产率,ee未测定).lcms:(m+1)m/z=324,326.化合物97的合成将dmf(0.2ml)中的(s)-5-(2-氯-8-氟-6-(1-氟乙基)-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-97-7(13.8mg,0.0426mmol)、n-(四氢-2h-吡喃-4-基)哌啶-4-胺4(9mg,0.047mmol)和dipea(8μl,0.047mmol)的混合物在120℃加热过夜。将反应混合物用盐水和etoac稀释。分离有机相,并用盐水(2x5ml)洗涤。将合并的有机相经na2so4干燥,并浓缩至干。将残留物经制备型-tlc(ch2cl2:meoh=96:4)纯化,得到化合物97,作为黄色固体物(5.0mg,25%产率,ee未测定).lcms:(m+1)m/z=472.int-97-6的合成将二氯(p-伞花烃)钌(ii)二聚体(1.13mg,1.8μmol)和(1s,2s)-(+)-n-p-甲苯磺酰基-1,2-二苯基乙二胺(1.63mg,4.4μmol)悬浮于水(0.74ml)中。将混合物用氮气脱气,然后在氮气气氛下,于70℃加热90min。将得到的混合物冷却至rt。加入酮int-97-5(120mg,0.37mmol)、甲酸钠(125.8mg,1.85mmol)和无水thf(0.37ml),并将反应物用氮气脱气。将反应混合物在40℃搅拌2.5h。将反应混合物用etoac稀释,并用盐水洗涤。将水相用etoac萃取。合并的有机层经na2so4干燥,并减压蒸发。将残留物经柱色谱(己烷:etoac=70:30)纯化,得到标题化合物,作为白色固体物(109mg,91%产率,ee未测定).lcms:(m+1)m/z=322,324.int-97-7的合成–78℃下,将n-(三甲基硅烷基)吗啉(92μl,0.52mmol)滴加到dast(67μl,0.508mmol)的干燥ch2cl2(0.2ml)溶液中。将得到的溶液在rt搅拌2.5h。将反应混合物在–78℃冷却,并滴加(s)-1-(2-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-6-基)乙-1-醇int-97-6(50mg,0.16mmol)的干燥ch2cl2(0.46ml)溶液。将反应混合物在rt搅拌过夜。然后,将反应混合物缓慢倒入10ml饱和nahco3溶液中。分离有机层,并将水相再用ch2cl2(3x10ml)萃取。将合并的有机相经na2so4干燥,并减压蒸发。将残留物经制备型-tlc(己烷:etoac=80:20)纯化,得到目标化合物,作为黄色油状物(20.5mg,41%产率,ee未测定).lcms:(m+1)m/z=324,326.化合物98的合成将dmf(0.2ml)中的(r)-5-(2-氯-8-氟-6-(1-氟乙基)-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-97-7(8.0mg,0.024mmol)、n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-97-4(6.8mg,0.036mmol)和dipea(6μl,0.036mmol)的混合物在120℃加热过夜。将反应混合物用盐水和etoac稀释。分离有机相,并用盐水(2x5ml)洗涤。将合并的有机相经na2so4干燥,并浓缩至干。将残留物经制备型-tlc(ch2cl2∶meoh=96∶4)纯化,得到化合物98,作为黄色固体物(2.6mg,23%产率)(ee未测定)。lcms:(m+1)m/z=472.实施例99-101反应试剂和条件:i)int-99-21(1.0equiv.),int-99-22(1.1equiv.),pph3(0.06equiv.),pdcl2(1/12equiv.),cs2co3(3.0equiv.),thf/h2o85℃,24h,69-72%;ii)int-99-23(1.0equiv.),bh3.me2s(2.06equiv.),h2o2,naoh,thf,50℃,2h,44-46%;iii)int-99-24(1.0equiv.),dast(1.1equiv),ch2cl2,0℃,2.5h;iv)int-99-25(1.0equiv.),10%aq.h2so4,thf,45℃,2h,54-57%(两步);v)int-99-26(1.0equiv.),胺(2.0equiv.),nabh(oac)3(2.0equiv.),acoh(2.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷,rt,过夜.int-99-23的合成将int-99-21(1.0当量)、乙烯基三氟硼酸钾(1.1当量)、pph3(0.06当量)、pdcl2(1/12当量)和cs2co3(3.0当量)在thf/h2o(9/1)中的混悬液在85℃加热24h。冷却至rt后,将粗品经硅藻土过滤。将滤液用etoac稀释,并用盐水洗涤,并将有机相减压浓缩。将残留物经柱色谱(己烷:etoac)纯化,得到int-99-23,作为浅黄色固体物。lcms:(m+1)m/z=411.int-99-24的合成在0℃,将bh3.me2s(2m,2.06当量)的thf溶液缓慢加入到int-99-23(1.0当量)的thf溶液中。将混合物升温至rt,并搅拌1h。将溶液冷却至0℃,并用30%h2o2水溶液(7当量)和naoh(1m,1.4当量)淬灭,并将混合物在50℃加热2h。将混合物减压浓缩,并将产物经柱色谱纯化(己烷:etoac)。lcms:(m+1)m/z=429.int-99-25的合成在0℃,向int-99-24(1.0当量)的ch2cl2水溶液中,缓慢加入dast(1.1当量)的ch2cl2溶液,并将反应物在0-5℃搅拌2.5h。将混合物用etoac稀释,并用盐水洗涤(2x)。将有机相用na2so4干燥,并减压浓缩。没有进一步纯化而使用粗产物(int-99-25和int-99-23的混合物)。lcms:(m+1)m/z=431.int-99-26的合成将int-99-25和int-99-23的thf/10%aq.h2so4溶液在45℃搅拌2h。冷却至rt后,将混合物用饱和naoh水溶液中和,并用etoac萃取产物。将有机相经na2so4干燥,并减压浓缩。将产物经柱色谱纯化(己烷/etoac),并得到酮int-99-26,作为浅黄色固体物(两步产率54-57%)。lcms:(m+1)m/z=387.化合物99、化合物100和化合物101的合成将1,2-二氯乙烷中的酮int-99-26(1.0当量)、适当的胺(2.0当量)、dipea(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在rt搅拌过夜。将混合物经硅藻土过滤,减压浓缩,并经hplc纯化,得到目标化合物。得到化合物99,作为灰白色固体物,产率91-92%(最后一步)。lcms:(m+1)m/z=472.得到化合物100,作为浅黄色固体物,产率62-64%(最后一步)。lcms:(m+1)m/z=472.得到化合物101,作为浅黄色固体物,产率81-83%(最后一步)。lcms:(m+1)m/z=458.实施例102反应试剂和条件:i)int-102-1(1.0equiv.),br2(1.2equiv.),fe(15mol%),nahco3(1.0equiv.),ch2cl2,50℃,过夜,88%;ii)int-102-2(1.0equiv.),nbuli(2.2equiv.),thf,-78℃,1h,随后dmf(2.0equiv.),-78℃~0℃,2h,11%;iii)int-102-3(1.0equiv.),氰基乙酸乙酯(2.0equiv.),nh4oac(5.0equiv.),1,4-二氧六环,95℃,过夜90%;iv)conc.hcl,1,4-二氧六环,100℃2h,83%;v)5(1.0equiv.),pocl3,110℃,2h,随后,n-羟基乙脒(1.2equiv.),dipea(3.0equiv.),1,4-二氧六环,110℃1.5h,35%;vi)int-102-6(1.0equiv.),1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0equiv.),dipea(2.0equiv.),iproh,110℃,过夜,62%;vii)10%aq.h2so4,thf,45℃,2h,90%;viii)int-102-8(1.0equiv.),胺(2.0equiv.),nabh(oac)3(3.0equiv.),acoh(3.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷,rt,过夜,73%-83%.int-102-2的合成向苯胺int-102-1(5.0g,27.9mmol)、铁粉(0.23g,4.2mmol,15mol%)和nahco3(2.34g,27.9mmol)的ch2cl2(100ml)混悬液中,滴加br2(1.73ml,33.5mmol)。将得到的混合物在50℃回流过夜。将混合物在2nnaoh和ch2cl2间进行分配。分离有机层,用na2so4干燥,并浓缩。将残留物经柱色谱纯化(己烷/etoac梯度),得到int-102-2,作为黄色固体物(6.35g,88%产率)。int-102-3的合成在-78℃,向int-102-2(6.35g,24.6mmol)的thf(30ml)溶液中,滴加2.5mnbuli的己烷溶液。在-78℃搅拌1h后,在相同温度下加入dmf(3.8ml,49.2mmol)。将温度升至0℃,并将混合物在0℃搅拌2h。然后,将混合物真空浓缩,将残留物经柱色谱纯化(己烷/etoac梯度),得到int-102-3,作为黄色固体物(0.56g,产率11%)。int-102-4的合成将1,4-二氧六环(5ml)中的int-102-3(560mg,2.68mmol)、氰基乙酸乙酯(605mg,5.36mmol)和nh4oac(1.03g,13.4mmol)的混合物在95℃搅拌并加热过夜。冷却至室温后,将混合物真空浓缩。将残留物在h2o和etoac间进行分配。分离有机层,用na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-102-4(浅黄色固体物)用于下一步反应(620mg,90%产率)。int-102-5的合成将化合物int-102-4(620mg,2.41mmol)在浓hcl(5ml)和1,4-二氧六环(5ml)中的混悬液在100℃加热2h。冷却至室温后,加入h2o(20ml)。将沉淀的产物过滤并干燥。没有进一步纯化,将产物int-102-5(浅黄色粉末)用于下一步反应(550mg,83%产率)。int-102-6的合成将酸int-102-5(550mg,2.0mmol)和pocl3(5ml)的混合物在110℃加热2h。冷却至室温后,真空除去过量的pocl3。将残留物溶解于1,4-二氧六环(5ml)中,并加入n-羟基乙脒(178mg,2.4mmol)和dipea(1.04ml,6.0mmol)。将得到的混合物在110℃加热1.5h。冷却至室温后,将混合物真空浓缩。将残留物利用15%etoac的己烷溶液用短硅胶通路纯化,得到int-102-6,作为浅黄色固体物(232mg,产率35%)。int-102-7的合成在室温,向int-102-6(184mg,0.55mmol)和dipea(0.19ml,1.1mmol)的iproh(3ml)溶液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(0.14ml,1.1mmol)。然后,将混合物在110℃加热过夜。冷却至室温后,将混合物浓缩,并经柱色谱纯化(己烷/etoac梯度),得到int-102-7,作为黄色固体物(150mg,产率62%)。lcms:(m+1)m/z=439.int-102-8的合成在室温下,向int-102-7(150mg,0.34mmol)的thf(2ml)溶液中,加入10%aq.h2so4(4ml)。然后,将混合物在45℃搅拌2h。冷却至rt后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(×2)。将合并的有机层经na2so4干燥,并真空浓缩。将残留物经柱色谱纯化(己烷/etoac梯度),得到int-102-8,作为黄色油状物(128mg,产率90%)。lcms:(m+1)m/z=395.化合物102的合成将1,2-二氯乙烷(1.5ml)中的int-102-8(9.0mg,0.023mmol)、(s)-3-氨基四氢吡喃(6.3mg,0.046mmol)和dipea(8μl,0.046mmol)的混合物在室温下搅拌10min。向所述混合物中,加入nabh(oac)3(14.5mg,0.069mmol)和acoh(4μl,0.069mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液真空浓缩。残留物经hplc(10-95%acn的h2o溶液,持续12min)纯化,得到化合物102,作为浅黄色固体物(8.0mg,73%产率)。lcms:(m+1)m/z=480.实施例103反应试剂和条件:i)int-103-10(1.0equiv.),n,0-二甲基羟基胺hcl(1.8equiv.),edcl(1.2equiv.),hobt(1.2equiv.),dipea(2.0equiv.),dmf,rt,18h,80%;ii)int-103-11(1.0equiv.),(cf3co)2o(1.2equiv.),tea(1.2equiv.),dcm,0℃-rt,5h,93%;iii)12(1.0equiv.),memgbr(4.0equiv.),thf,rt,6h,76%;iv)int-103-13(1.0equiv.),2nnaoh(1.7equiv),meoh,90℃,1.5h,99%;v)int-103-14(1.0equiv.),pylcl(1.0equiv.),meoh,100℃,18h,79%;vi)int-103-15(1.0equiv.),噁二唑酸(1.2equiv.),pocl3,110℃,1h;vii)int-103-16(1.0equiv.),1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2equiv.),dipea(1.2equiv.),etoh,120℃,18h,22%,两步;viii)int-103-17(1.0equiv.),fo2scf2co2me(5.0equiv.),cul(2.0equiv.),dmf,8%;ix)10%aq.h2so4,thf,50℃,1.5h,定量产率;x)int-103-19(1.0equiv.),胺(2.0equiv.),nabh(oac)3(3.0equiv.),acoh(3.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷,rt,18h,82%.int-103-11的合成将dmf(166ml)中的2-氨基-3-氟苯甲酸10(10.3g,66.4mmol)、n,o-二甲基羟基胺hcl(11.7g,119.5mmol)、edcl(15.3g,79.7mmol)、hobt(12.2g,79.7mmol)和dipea(23ml,132.8mmol)的混合物在室温搅拌过夜。将混合物用etoac稀释,并依次用1nnaoh、10%aq.hcl和盐水清洗。将有机层经na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-103-11(作为棕色油状物)用于下一步反应(10.48g,产率80%)。int-103-12的合成在0℃,向int-103-11(10.48g,52.9mmol)和tea(8.8ml,63.4mmol)的ch2cl2(200ml)溶液中,加入(cf3co)2o(8.8ml,63.4mmol)。将得到的混合物在室温搅拌5h。然后,将混合物用饱和nahco3水溶液和盐水清洗。将有机层经na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-103-12(作为棕色固体物)用于下一步反应(14.47g,产率93%)。lcms:(m+1)m/z=295.int-103-13的合成在0℃,向int-103-12(14.47g,49.2mmol)的无水thf(200ml)溶液中,加入3mmemgbr的乙醚(66ml,196.7mmol)溶液。将混合物在室温搅拌6h。然后,将混合物倒入碎冰中,以淬灭反应,并用etoac萃取。将有机层用10%aq.hcl和盐水清洗,na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-103-13(作为棕色油状物)用于下一步反应(9.36g,产率76%)。int-103-14的合成在室温下,向int-103-13(9.36g,37.5mmol)的meoh(32ml)溶液中,加入2nnaoh。然后,将混合物在90℃加热1.5h。冷却至室温后,将混合物在盐水和ch2cl2之间进行分配。将水层用ch2cl2萃取。将合并的有机萃取物用na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-103-14(作为深褐色油状物)用于下一步反应(5.70g,产率99%)。int-103-15的合成将meoh(125ml)中的int-103-14(5.7g,37.2mmol)和pyicl(9.0g,37.2mmol)的混合物在100℃加热过夜。冷却至室温后,将混合物真空浓缩。残留物经柱色谱纯化(己烷/etoac梯度),得到int-103-15,作为黄色固体物(7.91g,76%产率)。lcms:(m+1)m/z=280.int-103-16的合成将int-103-15(2.20g,7.9mmol)、二唑酸(1.34g,9.5mmol)和pocl3(10ml)的混合物在110℃搅拌1h。冷却至室温后,真空除去过量的pocl3。将残留物在h2o和ch2cl2之间进行分配。将有机层分离,用饱和nahco3水溶液清洗,用na2so4干燥,并浓缩。没有进一步纯化,将残留物int-103-16用于下一步反应。int-103-17的合成在室温下,向粗品int-103-16(2.70g,6.7mmol)和dipea(1.4ml,8.0mmol)在etoh(50ml)的浆状物中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.0ml,8.0mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物真空浓缩,并将残留物经柱色谱(己烷/etoac梯度)纯化,得到int-103-17,作为棕色固体物(0.75g,两步产率22%)。lcms:(m+1)m/z=511.int-103-18的合成在室温下,向17(200mg,0.39mmol)和cui(148mg,0.78mmol)在dmf(4ml)中的浆状物中,加入氟磺酰基二氟乙酸甲酯(0.24ml,1.95mmol)。然后,将混合物在80℃加热1h。经硅藻土过滤后,浓缩滤液。将残留物经hplc(50-95%acn的h2o溶液,15min)纯化,得到int-103-18,作为橙色固体物(14.8mg,产率8%)。lcms:(m+1)m/z=453.int-103-19的合成在室温下,向int-103-8(14.8g,0.033mmol)的thf(1ml)溶液中,加入10%aq.h2so4(2ml)。然后,将混合物在50℃搅拌1.5h。冷却至rt后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(×2)。将合并的有机层经na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-103-19(作为黄色固体物)用于下一步反应。(13.3mg,定量产率)。lcms:(m+1)m/z=409.化合物103的合成将1,2-二氯乙烷(1.5ml)中的int-103-19(13.3mg,0.033mmol)、4-氨基四氢吡喃(6.6mg,0.066mmol)和dipea(11μl,0.066mmol)的混合物在rt下搅拌10min。向所述混合物中,加入nabh(oac)3(20.8mg,0.099mmol)和acoh(6μl,0.099mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液真空浓缩。残留物经hplc(10-95%acn的h2o溶液,持续12min)纯化,得到化合物103,作为浅橙色固体物(13.2mg,产率82%)。lcms:(m+1)m/z=494.实施例104-107根据上文公开的化合物103的方法,得到化合物104-107。以定量产率(最后一步),得到化合物104,作为浅黄色固体物。lcms:(m+1)m/z=494.以定量产率(最后一步),得到化合物105,作为浅黄色固体物。lcms:(m+1)m/z=494.以97%产率(最后一步),得到化合物106,作为浅黄色固体物。lcms:(m+1)m/z=480.以84%产率(最后一步),得到化合物107,作为浅黄色固体物。lcms:(m+1)m/z=480.实施例108-109反应试剂和条件:i)int-36-4(1.0equiv.),memgbr(4.0equiv.),thf,0℃tort,2.5h,64-66%;ii)int-108-25(1.0equiv.),naoh(1.7equiv.),meoh∶h2o1∶1,90℃,3h,90-93%;iii)int-103-2(1.0equiv.),bh3·thf(3.0equiv.),thf,rt,18h;iv)27(1.0equiv.),mno2(6.0equiv.),ch2cl2,rt,过夜,69-71%,两步;v)b2pin2(1.2equiv.),pd(dppf)cl2·ch2cl2(0.1equiv.),koac(3.0equiv.),二氧六环80℃,18h,63-65%;vi)cf3ch2i(2.0equiv.),pd2(dba)3(0.1equiv.),xantphos(0.2equiv.),cs2co3(4.0equiv.),二氧六环,h2o(1.8equiv.),80℃,18h;vii)9(1.0equiv.),pocl3,110℃,1.5h;viii)1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0equiv.),dipea(2.0equiv.),etoh,110℃,过夜;ix)10%aq.h2so4,thf,45℃,2h;x)4-氨基四氢吡喃(2.0equiv.),nabh(oac)3(2.0equiv.),acoh(2.0equiv.),1,2-二氯乙烷,rt,18h.int-108-26的合成在0℃,向int-36-4(1.0当量)的thf(200ml)溶液中,加入1.4mmemgbr(4.0当量)的thf:甲苯(1:3)溶液,并将得到的混合物在室温搅拌2.5h。然后,将反应物倒入冰中,用2mhcl酸化至ph2,并用etoac(2x)萃取。将合并的有机层经na2so4干燥,并减压浓缩,得到25,作为黄色油状物(产率64-66%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=325,327.向int-108-25(1.0当量)的meoh溶液中,加入2mnaoh(1.7当量)水溶液,并将反应物在90℃加热3h。将反应混合物用水稀释,并经过滤收集固体物,得到int-108-26,作为黄色固体物(产率90-93%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=231,233.int-108-28的合成在0℃下,向int-108-2(1.0当量)的thf(60ml)溶液中滴加bh3.thf配合物(3.0当量)的溶液。然后将混合物在室温搅拌18h。将反应在0℃下用meoh淬灭,浓缩,重悬于etoac中,并用饱和nahco3水溶液洗涤。有机相经na2so4干燥并浓缩,得到醇int-108-27,其无需进一步纯化即可用于下一步。lcms:(m+1)m/z=219,221。向醇27的ch2cl2溶液中,缓慢加入活化的mno2(6.0当量),并将混合物在室温下搅拌过夜。将混合物经硅藻土过滤,并减压除去溶剂,得到醛int-108-28,作为黄色固体物(两步产率69-71%),没有进一步纯化,将其用于下一步骤。lcms:(m+23)m/z=239,241.int-108-29和int-108-30的合成将二氧六环中的int-108-28(1.0当量)、双(频哪醇基)二硼(1.2当量)、pd(dppf)cl2·ch2cl2(0.1当量)和koac(3.0当量)的混合物在80℃加热18h。将混合物冷却至室温,经硅藻土过滤,减压浓缩,并经柱色谱纯化(己烷/etoac),得到硼酸酯int-108-30,作为灰白色固体物(产率63-65%)。lcms:(m+1)m/z=266.利用相同反应条件,以93-95%产率得到硼酸酯int-108-29,作为灰色固体物。lcms:(m+1)m/z=280.int-108-31和int-108-32的合成向密封瓶中的pd2(dba)3(0.1当量)、xantphos(0.2当量)和cs2co3(4.0当量)的二氧六环混悬液中,加入硼酸酯int-108-30(1.0当量)和cf3ch2i(2.0当量)的二氧六环溶液。将反应混合物在室温搅拌1分钟,然后加入h2o(1.8当量)。将混合物在80℃搅拌18h。冷却后,将反应混合物用etoac稀释,并用水和盐水洗涤。将有机层经na2so4干燥,浓缩,并经柱色谱(己烷/etoac)纯化,得到int-108-32,作为橙色固体物(产率95-97%)。lcms:(m+1)m/z=222.用相同方式,以77-79%产率得到酮int-108-31,作为浅黄色固体物。lcms:(m+1)m/z=236.int-108-33和int-108-34的合成将醛int-108-32(1.0当量)、酸int-108-9(1.0当量)和pocl3的混合物在110℃搅拌1.5h。减压除去过量的pocl3。将残留物再悬浮于饱和nahco3水溶液中,并用etoac(2x)萃取。将合并的有机层经na2so4干燥,并浓缩至干。将粗产物经柱色谱(己烷/etoac)纯化,得到喹啉int-108-34,作为白色固体物(产率15-17%).lcms:(m+1)m/z=346.用相同方式,以28-30%产率得到喹啉int-108-33,作为浅灰色固体物。lcms:(m+1)m/z=360.int-108-35和int-108-36的合成将int-108-34(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0当量)和dipea(2.0当量)的混合物在110℃加热过夜。冷却至室温后,将混合物减压浓缩,并经柱色谱纯化(己烷/etoac),得到缩酮36,作为黄色固体物(产率61-64%)。lcms:(m+1)m/z=453.用类似方式,以84-86%产率得到缩酮int-108-35,作为浅黄色固体物。lcms:(m+1)m/z=467.int-108-37和int-108-38的合成在室温下,向缩酮int-108-36(1.0当量)的thf溶液中,加入10%aq.h2so4。然后,将混合物在45℃搅拌2h。冷却至rt后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(2×)。将合并的有机层经na2so4干燥,并浓缩,并经制备型-tlc(己烷:etoac7:3)纯化,得到酮int-108-38,作为黄色固体物(产率54-56%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=409.用类似方式,以88-90%产率得到酮int-108-37,作为浅黄色固体物。lcms:(m+1)m/z=423.化合物108和化合物109的合成将1,2-二氯乙烷中的酮int-108-38(1.0当量)、4-氨基四氢吡喃(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在室温搅拌18h。将混合物减压浓缩,并经制备型-tlc(etoac:iproh,95:5)纯化,得到化合物108,作为黄色固体物(产率19-21%)。lcms:(m+1)m/z=494.用类似方式,以22-24%产率得到化合物109,作为黄色固体物。lcms:(m+1)m/z=508.实施例110根据化合物109的方法,从酮int-108-37和相应的胺开始,以产率49-51%得到化合物110,作为浅黄色固体物。lcms:(m+1)m/z=508.实施例111-115反应试剂和条件:i)int-111-1(1.0equiv.),b2pin2(1.2equiv.),pd(dppf)cl2-ch2cl2(0.03equiv.),koac(3.0equiv.),1,4-二氧六环90℃,过夜,55%;ii)int-111-2(1.0equiv.),cf3ch2l(2.0equiv.),pd2(dba)2(0.1equiv.),xantphos(0.1equiv.),cs2co3(4equiv.),h2o(2.0equiv),1,4-二氧六环80℃,过夜,78%;iii)int-111-3(1.0equiv.),pylcl(1.05equiv.),meoh,90℃,过夜,67%;iv)4(1.0equiv),噁二唑酸(1.2equiv.),pocl3,110℃,1h;v)int-111-5(1.0equiv.),1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2equiv.),dipea(2.0equiv.),etoh,120℃,过夜,26%,两步);vi)int-111-6(1.0equiv.),zn(cn)2(1.0equiv.),zn(0.1equiv.),pd(pph3)4(0.1equiv.),thf/dmf,80℃,过夜,84%;vii)10%aq.h2so4,thf,50℃,2h,定量产率;viii)int-111-8(1.0equiv.),胺(2.0equiv.),nabh(oac)3(3.0equiv.),acoh(3.0equiv.),dipea(2.0equiv.),1,2-二氯乙烷过夜,89%~定量产率int-111-2的合成将1,4-二氧六环(30ml)中的int-111-1(6.42g,30.0mmol)、双(频哪醇基)二硼(9.14g,36.0mmol)、[1,1′-双(二苯基膦基)二茂铁]二氯化钯(ii)·ch2cl2(0.73g,0.9mmol)和koac(8.82g,90.0mmol)的混合物在90℃搅拌过夜。冷却至室温后,将混合物过滤,并将滤液减压浓缩。将残留物经柱色谱(己烷/etoac梯度),得到int-111-2,作为浅黄色固体物(4.31g,产率55%)。int-111-3的合成将1,4-二氧六环(32ml)中的int-111-2(1.70g,6.51mmol)、pd2(dba)2(0.62g,0.65mmol)、xantphos(0.38g,0.65mmol)、cs2co3(8.50g,26.04mmol)和cf3ch2i(1.3ml,13.02mmol)的混合物在室温搅拌1min。向其中,加入h2o(0.23ml,13.02mmol),并将得到的混合物在密封瓶中于80℃搅拌过夜。冷却至室温后,将混合物经硅藻土过滤,并将滤液减压浓缩。将残留物经柱色谱纯化(己烷/etoac梯度),得到int-111-3,作为黄色固体物(1.11g,产率78%)。lcms:(m+1)m/z=218.int-111-4的合成将meoh(20ml)中的int-111-3(1.11g,5.11mmol)和pyicl1(1.30g,5.36mmol)的混合物在90℃搅拌过夜。冷却至室温后,将混合物减压浓缩。将残留物经柱色谱(己烷/etoac梯度)纯化,得到int-111-4,作为黄色固体物(1.17g,产率67%)。lcms:(m+1)m/z=344.int-111-5的合成将int-111-4(1.17g,3.41mmol)、二唑酸(0.58g,4.09mmol)和pocl3(7ml)的混合物在110℃搅拌1h。冷却至至温后,减压除去过重的pocl3。在0℃,向所述残留物中,加入h2o,并将混合物在0℃搅拌10min。将沉淀的粗品氯喹啉5过滤,用h2o清洗,并减压干燥。没有进一步纯化,将粗品int-111-5用于下一步反应。int-111-6的合成在室温,向粗品氯喹啉int-111-5(1.59g,3.41mmol)和dipea(1.2ml,6.82mmol)的etoh混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(4.09ml,7.9mmol)。然后,将所述混合物在120℃搅拌过夜。冷却至室温后,将混合物浓缩,并经柱色谱纯化(己烷/etoac梯度),得到int-111-6,作为深褐色泡沫状物(513mg,两步产率26%)。lcms:(m+1)m/z=575.int-111-7的合成将int-111-6(513mg,0.89mmol)、zn(cn)2(105mg,0.89mmol)、zn(6mg,0.09mmol)和pd(pph3)4(103mg,0.09mmol)在thf/dmf(12ml,1:1(v/v))中的混合物于80℃搅拌过夜。冷却至室温后,将混合物过滤,并减压浓缩滤液。将残留物经柱色谱(己烷/etoac梯度)纯化,得到int-111-7,作为黄色固体物(356mg,产率84%)。lcms:(m+1)m/z=474.int-111-8的合成在室温下,向int-111-7(356mg,0.75mmol)的thf(3ml)溶液中,加入10%aq.h2so4(6ml)。然后,将混合物在50℃搅拌2h。冷却至rt后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(×2)。将合并的有机萃取物经na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-111-8(作为黄色固体物)用于下一反应(322mg,定量产率)。lcms:(m+1)m/z=430.化合物111、化合物112、化合物113、化合物114、化合物115的合成将1,2-二氯乙烷(1.5ml)中的int-111-8(11mg,0.026mmol)、适当的胺(0.052mmol)和dipea(9μl,0.052mmol)的混合物在室温搅拌10min。向所述混合物中,加入nabh(oac)3(16.3mg,0.078mmol)和acoh(5μl,0.078mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。残留物经hplc(10-95%acn的h2o溶液,持续12min)纯化后,得到标题化合物:化合物111,11.8mg,黄色油状物,产率89%。lcms:(m+1)m/z=515;化合物112,13.2mg,黄色固体物,产率89%。lcms:(m+1)m/z=515;化合物113,13.2mg,黄色固体物,产率89%。lcms:(m+1)m/z=515;化合物114,12.8mg,黄色固体物,产率89%。lcms:(m+1)m/z=501;和化合物115,12.8mg,黄色固体物,产率89%。lcms:(m+1)m/z=501.实施例116-117化合物116的合成向化合物30(30mg,0.060mmol)和三丁基(丙-1-炔-1-基)锡烷(78mg,0.24mmol)的1,4-二氧六环(1.5ml)溶液中,加入双(三苯基膦)氯化钯(ii)(3.3mg,4.8μmol),并将反应物在氮气气氛下于100℃微波辐射45min。将混合物过滤,并将溶液减压浓缩。将残留物经制备型-hplc纯化,得到化合物16(5.0mg,产率18%),作为白色固体物。lcms:(m+1)m/z=464;保留时间:2.50min(方法1).化合物117的合成化合物117的制备与化合物116相同,除了用(r)-1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-3-基)哌啶-4-胺代替1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺化合物30。lcms:(m+1)m/z=464;保留时间:2.55min(方法1).实施例118反应试剂和条件:i)int-118-14(1.0equiv.),nbs(1.0equiv.),ch2cl2,rt,18h,84-86%;ii)int-118-15(1.0equiv.),(meo)nh(me).hci(1.8equiv.),edcl(1.2equiv.),hobt(1.2equiv.),dipea(2.0equiv.),dmf,rt,4h,84-86%;iii)int-118-16(1.0equiv.),(cf3co)2o(1.3equiv.),tea(1.2equiv.),ch2cl2,0℃tort,过夜,90-92%;iv)int-118-17(1.0equiv.),bet3(30equiv.),cs2co3(3.0equiv),pd(dppf)cl2(0.02equiv.),thf,70℃,1.5h,45-47%;v)int-118-18(1.0equiv.),memgbr(5.0equiv.),thf,0℃tort,2.5h,85-87%;vi)int-118-19(1.0equiv.),naoh(1.7equiv.),meoh∶h2o1∶1,90℃,3h,80-82%;vii)int-118-20(1.0equiv.),8(2.0equiv.),p-tsoh(cat),150℃,2h,19-21%;viii)21,pocl3,110℃,2h,97-98%;ix)int-118-22(1.0equiv.),1,4-二氧杂-8-氮杂螺[4.5]癸烷(2.0equiv.),dipea(2.0equiv.),etoh,110℃,过夜,73-75%;x)10%aq.h2so4,thf,45℃,2h,83-85%;xi)int-118-24(1.0equiv.),4-氨基四氢吡喃(1.5equiv.),nabh(oac)3(2.0equiv.),acoh(2.0equiv.),1,2-二氯乙烷,rt,过夜,89-90%.int-118-2的合成向2-氨基-3-氟苯甲酸int-118-1(15.0g,96.7mmol)的ch2cl2(250ml)混悬液中,加入n-碘代琥珀酰亚胺(17.2g,96.7mmol),并将混合物在室温搅拌过夜。将产物过滤,用ch2cl2清洗,并减压干燥,得到int-118-2,作为浅褐色固体物(24.7g,产率91%)。int-118-3的合成将dmf(166ml)中的int-118-2(6.2g,22.06mmol)、n,o-二甲基羟基胺hcl(3.9g,39.71mmol)、edcl(5.1g,26.47mmol)、hobt(4.1g,26.47mmol)和dipea(7.7ml,44.12mmol)的混合物在室温搅拌过夜。将混合物用etoac稀释,并依次用1nnaoh、10%aq.hcl和盐水清洗。将有机层经na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-118-3(作为棕色油状物)用于下一步反应(6.9g,产率96%)。int-118-4的合成在0℃,向int-118-3(6.9g,21.3mmol)和tea(3.6ml,25.6mmol)的ch2cl2(70ml)溶液中,加入(cf3co)2o(3.9ml,27.7mmol)。将得到的混合物在室温搅拌过夜。然后,将混合物用饱和nahco3水溶液和盐水洗涤。将有机层经na2so4干燥,并蒸发至干。没有进一步纯化,将产物int-118-4(作为棕色油状物)用于下一步反应(8.8g,产率98%)。lcms:(m+1)m/z=421.int-118-5的合成在0℃,向int-118-4(4.47g,10.6mmol)的无水thf(100ml)溶液中,加入1.4mmemgbr的乙醚溶液(42ml,59.4mmol)。将混合物在室温搅拌18h。然后,将混合物倒入碎冰中,以淬灭反应,并用etoac萃取。将有机层用10%aq.hcl和盐水洗涤,na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-118-5(作为深橙色油状物)用于下一步反应(1.68g,产率60%)。int-118-6的合成在室温下,向int-118-5(1.22g,4.63mmol)的meoh(7ml)溶液中,加入2nnaoh(4ml)。然后,将混合物在90℃加热1.5h。冷却至室温后,将混合物在盐水和ch2cl2之间进行分配。将水层用ch2cl2萃取。将合并的有机萃取物用na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-118-6(作为黄色固体物)用于下一步反应(0.73g,产率94%)。int-118-7的合成将int-118-6(371mg,2.22mmol)、二唑酸(380mg,2.67mmol)和pocl3(4.5ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。将残留物在ch2cl2和h2o之间进行分配。分离有机层,用饱和nahco3水溶液洗涤,用na2so4干燥,并浓缩。残留物经柱色谱(己烷/etoac梯度)纯化,得到化合物int-118-7,作为浅褐色固体物(0.25g,产率39%)。lcms:(m+1)m/z=292.化合物118的合成将dmf(1ml)中的int-118-7(30mg,0.103mmol)、胺(127mg,0.309mmol)和2,2,6,6-四甲基哌啶(18μl,0.103mmol)的混合物在150℃加热过夜。冷却至室温后,将混合物浓缩。将残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物118,作为浅黄色固体物(23mg,产率51%)。lcms:(m+1)m/z=440.实施例119-120int-119-12的合成向8-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷(int-83-2,100mg,0.22mmol)和双(三苯基膦)氯化钯(ii)(14mg,18umol)的二氧六环(5ml)混悬液中,加入二甲基锌(10%wt的己烷溶液,0.50ml),并将反应物在60℃搅拌1h。将反应物用甲醇和柠檬酸溶液(5%aq.)淬灭,并用乙酸乙酯萃取两次。将有机层合并,用盐水洗涤,经na2so4干燥,并减压浓缩。将残留物经硅胶色谱纯化,得到8-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷(int-119-12),作为黄色固体物(42mg,产率49%)。lcms(esi):m/z399(m+h);保留时间:3.05min(方法1).int-119-13的合成利用用于1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮(int-30-11f)的通用方法,用8-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷(int-119-12)代替8-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷(int-1-1d),合成int-119-13。lcms(esi):m/z355(m+h);保留时间:2.60min(方法1).化合物119的合成方法与(r)-1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-3-基)哌啶-4-胺(化合物31)相同,但用1-(8-氟-4,6--二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮(int-119-13)代替1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)哌啶-4-酮(int-30-11f),得到化合物119。lcms(esi):m/z440(m+h);保留时间:2.41min(方法1).化合物120的合成方法与化合物119的相同,但用(s)-四氢-2h-吡喃-3-胺盐酸盐(2.0当量)代替(r)-四氢-2h-吡喃-3-胺盐酸盐(2.0当量),得到化合物120。lcms(esi):m/z440(m+h);保留时间:2.44min(方法1).实施例121-133利用具有适当立体化学的胺和适当的酮,用下文公开的化合物134-137的方式,得到化合物121-128。用下文公开的化合物147-152的方式或者上文公开的化合物67-71的方式,得到化合物129-133。实施例134-137反应试剂和条件.:i)int-134-44(1.0equiv.),int-134-45(2.0equiv.),nabh(oac)3(2.0equiv.),acoh(2.0equiv.),1,2-二氯乙烷,rt,过夜;ii)tfa(10equiv.),ch2cl2,rt,2h;iii)int-134-46(2.0equiv.),int-134-47(1.0equiv.),dipea(4.0equiv.),etoh,110℃,过夜,65-68%(3步).int-134-46的合成将dce中的int-134-44(1.0当量)、int-134-45(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在rt搅拌过夜。将混合物用饱和nahco3水溶液淬灭,并将产物用ch2cl2(3x)萃取。将有机相用na2so4干燥,并减压浓缩。没有进一步纯化而使用产物。lcms:(m+1)m/z=303.将ch2cl2中的上述产物和tfa(10.0当量)的混合物在rt搅拌2h。将混合物减压浓缩,并且不需纯化而使用产物。lcms:(m+1)m/z=203.化合物134的合成将etoh中的int-134-46(2.0当量)、int-134-47(1.0当量)和dipea(4.0当量)混合物在110℃加热过夜。减压浓缩混合物,并经制备型-tlc(ch2cl2:meoh=96:4)纯化产物,以65-68%产率(3步)得到化合物134。化合物135、化合物136和化合物137的合成除了用适当的立体异构体代替原料int-134-44,用与化合物134相同的方式,制备化合物135、化合物136和化合物137。化合物135,浅灰色固体物;化合物136,浅黄色固体物,化合物137,浅灰色固体物。实施例138试剂和条件:i)int-138-1(1.0当量),nis(1.0当量),ch2cl2,rt,6h,76%;ii)int-138-2(1.0当量),bh3-thf(5.0当量),thf,rt,6h,定量产率;iii)int-138-3(1.0当量),mno2(6.0当量),ch2cl2,rt,过夜,定量产率;iv)int-138-4(1.0当量),zn(cn)2(1.0当量),zn(0.1当量),pd(pph3)4(0.05当量),dmf,80℃,过夜,42%;v)int-138-5(1.0当量),二唑酸(1.2当量),pocl3,110℃,1h,92%;vi)int-138-6(1.0当量),胺(2.0当量),dipea(2.0当量),ch3cn,120℃,过夜,17%.int-138-2的合成向2-氨基-3-氯苯甲酸int-138-1(17.16g,100mmol)的ch2cl2(250ml)混悬液中,加入n-碘代琥珀酰亚胺(17.80g,100mmol),并将混合物在室温搅拌。将产物过滤,用ch2cl2洗涤,并减压干燥。没有进一步纯化,将产物int-138-2(作为灰白色固体物)用于下一步反应(22.61g,产率76%)。lcms:(m-1)m/z=296.int-138-3的合成在0℃,向int-138-2(22.61g,76.0mmol)的thf(100ml)混悬液中,滴加bh3.thf络合物溶液(1m的thf溶液,380ml,380mmol)。然后,将混合物在室温搅拌6h。通过在0℃缓慢加入meoh而淬灭反应,并将混合物浓缩至干。没有进一步纯化,将产物int-138-3(作为粉色固体物)用于下一步反应(21.54g,定量产率)。lcms:(m-1)m/z=282.int-138-4的合成将ch2cl2(500ml)中的int-138-3(21.54g,76.0mmol)和活化mno2(40.0g,456.0mmol)的混合物在室温搅拌过夜。将反应混合物经硅藻土过滤,得到int-138-4,作为黄色固体物(21.39g,定量产率)。int-138-5的合成将dmf(70ml)中的int-138-4(9.95g,35.35mmol)、zn(cn)2(4.15g,35.35mmol)、zn粉(0.23g,3.54mmol)和pd(pph3)4(2.04g,1.77mmol)的混合物于80℃加热过夜。冷却至室温后,将混合物经硅藻土过滤,并浓缩滤液。将残留物经柱色谱(己烷/etoac)纯化,得到int-138-5,作为浅黄色固体物(2.68g,产率42%)。lcms:(m+1)m/z=181.int-138-6的合成将int-138-5(180mg,1.00mmol)、二唑酸(175mg,1.23mmol)和pocl3(3ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃,向残留物中加入水。将混合物在etoac和盐水之间进行分配。分离水层,并用etoac(×2)萃取。将合并的有机层用na2so4干燥,并浓缩至干。没有进一步纯化,将产物int-138-6(作为棕色固体物)用于下一步反应(281mg,产率92%)。化合物138的合成将ch3cn(1ml)中的int-138-6(17mg,0.056mmol)、胺(33mg,0.112mmol)和dipea(19μl,0.112mmol)的混合物在120℃加热过夜。冷却至室温后,将混合物浓缩。残留物经制备型-tlc(ch2cl2:meoh=95:5~90:10)纯化,得到化合物138,作为黄色油状物(4.3mg,产率17%)。lcms:(m+1)m/z=453.实施例139int-139-28的合成向2-氨基-3-氟苯甲酸25(15.0g,96.7mmol)的ch2cl2(250ml)混悬液中,加入n-碘代琥珀酰亚胺(17.2g,96.7mmol)。将混合物在室温搅拌过夜。将产物过滤,用ch2cl2洗涤,并减压干燥,得到碘代苯甲酸26,作为灰白色固体物。在0℃,向int-139-int-139-26(22.48g,80.0mmol)的thf(200ml)溶液中,滴加bh3.thf络合物(1mthf溶液,400ml,400mmol)溶液。然后,将混合物在室温搅拌过夜。在0℃,通过缓慢加入meoh而淬灭反应。将溶剂除去至干燥后,将粗醇int-139-int-139-27溶解于ch2cl2(500ml)中,并加入活化的mno2(41.7g,480mmol)。将混合物在室温搅拌过夜。将粗产物用短硅胶通路(己烷:etoac,1:1)纯化,得到醛int-139-int-139-28,作为黄色固体物(11.4g,历经三步的产率45%)。lcms:(m-1)m/z=264.int-139-int-139-30的合成将醛int-139-int-139-28(5.74g,21.6mmol)、二唑酸int-139-int-139-12(3.69g,26.0mmol)和pocl3(22ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃,向所述残留物中加入水,并将混合物在0℃搅拌10min。将粗氯化物29过滤,用h2o洗涤,并减压干燥。在室温,向29在etoh(100ml)中的混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(5.5ml,43.2mmol)和dipea(7.5ml,43.2mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩并经柱色谱纯化(己烷/etoac梯度),得到喹啉int-139-int-139-30,作为黄色油状物(2.50g,两步产率23%).lcms:(m+1)m/z=497.int-139-31的合成将int-139-30(2.50g,5.04mmol)、zn(cn)2(0.59g,5.04mmol)、zn粉(33mg,0.50mmol)和pd(pph3)4(0.58g,0.50mmol)在thf/dmf(50ml,1:1(v/v))中的混合物于80℃加热5h。冷却至室温后,将混合物经硅藻土过滤,并浓缩滤液。将残留物经柱色谱(己烷/etoac梯度)纯化,得到氰基喹啉int-139-31,作为黄色油状物(0.93g,产率47%)。lcms:(m+1)m/z=396.int-139-32的合成向int-139-31(0.47g,1.19mmol)的thf(5ml)溶液中,加入10%aq.h2so4(12ml)。将混合物在45℃搅拌4h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(2×40ml)。将合并的有机层经na2so4干燥,并浓缩至干。经柱色谱纯化(己烷/etoac梯度),得到酮int-139-int-139-32,作为黄色固体物(308mg,产率74%)。lcms:(m+1)m/z=352.化合物139的合成将1,2-二氯乙烷(1.5ml)中的int-139-32(15.8mg,0.066mmol)、(s)-3-氨基四氢吡喃盐酸盐(12.4mg,0.09mmol)和dipea(16μl,0.08mmol)的混合物在室温下搅拌10min。向所述混合物中,加入nabh(oac)3(28.6mg,0.135mmol)和acoh(8μl,0.135mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物139,作为黄色油状物(18.1mg,产率92%)。lcms:(m+1)m/z=437.实施例140int-140-36的合成向2-氨基苯甲酸33(6.6g,48.1mmol)的ch2cl2(150ml)混悬液中,加入n-碘代琥珀酰亚胺(17.1g,96.2mmol)。将混合物在室温搅拌过夜。将产物过滤,用ch2cl2洗涤,并减压干燥,得到碘代苯甲酸int-140-34,作为灰白色固体物。在0℃,向int-140-34(12.53g,32.2mmol)的thf(100ml)溶液中,滴加bh3.thf络合物(1mthf溶液,161ml,161mmol)溶液。然后,将混合物在室温搅拌过夜。在0℃,通过缓慢加入meoh而淬灭反应。将溶剂除去至干燥后,将粗醇int-140-35溶解于ch2cl2(200ml)中,并加入活化的mno2(16.8g,193.2mmol)。将混合物在室温搅拌过夜。将粗产物用短硅胶通路纯化,得到醛int-140-36,作为黄色固体物(5.83g,历经三步的产率32%)。lcms:(m-1)m/z=374.int-140-38的合成将醛int-140-36(4.81g,12.9mmol)、二唑酸12(3.69g,15.5mmol)和pocl3(13ml)的混合物在110℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃,向所述残留物中加入水。将混合物在0℃搅拌10min。将粗氯化物int-140-37过滤,用h2o洗涤,并减压干燥。在室温,向int-140-37在etoh(65ml)中的混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(3.3ml,25.8mmol)和dipea(4.5ml,25.8mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩并经柱色谱纯化(己烷/etoac梯度),得到喹啉int-140-38,作为黄色油状物(1.56g,两步产率20%).lcms:(m+1)m/z=605.int-140-39的合成将int-140-38(1.56g,2.58mmol)、zn(cn)2(0.61g,5.16mmol)、zn粉(34mg,0.52mmol)和pd(pph3)4(0.60g,0.52mmol)在thf/dmf(30ml,1:1(v/v))中的混合物于80℃加热5h。冷却至室温后,将混合物经硅藻土过滤,并浓缩滤液。将残留物经柱色谱(己烷/etoac梯度)纯化,得到氰基喹啉int-140-39,作为黄色油状物(0.62g,产率60%)。lcms:(m+1)m/z=403.int-140-40的合成向int-140-39(0.57g,1.42mmol)的thf(5ml)溶液中,加入10%aq.h2so4(12ml)。将混合物在45℃搅拌4h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(2×40ml)。将合并的有机层经na2so4干燥,并浓缩至干。没有进一步纯化,将酮int-140-40(作为橙色固体物)用于下一步反应(513mg,定量产率)。lcms:(m+1)m/z=359.化合物140的合成将1,2-二氯乙烷(1.5ml)中的int-140-40(13.0mg,0.036mmol)、(s)-3-氨基四氢吡喃盐酸盐(10.0mg,0.072mmol)和dipea(13μl,0.072mmol)的混合物在室温下搅拌10min。向所述混合物中,加入nabh(oac)3(23.0mg,0.108mmol)和acoh(6μl,0.108mmol)。将得到的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。残留物经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物140,作为黄色油状物(14.1mg,产率88%)。lcms:(m+1)m/z=444.实施例141int-141-2的合成向int-141-1(2.04g,9.38mmol)的etoh(14ml)溶液中,加入丙二酸二乙酯(1.99ml,13.1mmol)和催化量哌啶。将反应混合物回流24h。将反应混合物冷却至rt,并将得到的固体物经真空过滤收集,用etoh洗涤,得到int-141-2,作为白色固体物(2.6g,产率88%)。lcms:(m-1)m/z=313.int-141-3的合成将int-141-2(806mg,2.56mmol)和氯氧化磷(v)(5ml)的混合物在110℃加热1h。减压除去氯氧化磷(v)。向残留物中加入水,并经真空过滤收集沉淀物,得到int-141-3,作为白色固体物(850mg,定量产率)。int-141-4的合成向int-141-3(720mg,2.16mmol)的thf(3.5ml)溶液中,加入naoh(519mg,13.0mmol)和水(7ml)。将反应物在rt搅拌2h。将溶剂减压蒸发,然后加入树脂amberliteirn77和meoh(15ml)。将得到的混合物在rt搅拌15min,然后滤掉树脂。将滤液浓缩至干,得到int-141-4,作为灰白色固体物(660mg,定量产率)。lcms:(m-1)m/z=303.int-141-5的合成将二氯甲烷(4ml)中的int-141-4(660mg,2.16mmol)和n,n-二甲基甲酰胺(0.2ml)的混合物用草酰氯(2m的二氯甲烷溶液,4ml)处理。将反应混合物在rt搅拌2h。将反应混合物减压蒸发。将残留物溶解于二氯甲烷(4ml)和n,n-二甲基甲酰胺(4ml)中,然后用n-羟基乙酰胺(160mg,2.16mmol)和dipea(1.13ml,6.48mmol)处理。将反应混合物在120℃加热2h,然后冷却至rt。真空除去二氯甲烷。向残留物中加入etoac和水。分离有机相,避光用盐水洗涤(3x20ml)。将有机相经na2so4干燥,并浓缩至干,随后经柱色谱(己烷:etoac=92:8),得到int-141-5,作为黄色固体物(178mg,产率24%).lcms:(m+1)m/z=342,344.int-141-6的合成将etoh(3ml)中的int-141-5(150mg,0.44mmol)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(0.11ml,0.88mmol)和dipea(0.15ml,0.88mmol)的混合物在120℃微波下加热50min。将反应混合物浓缩,并经柱色谱纯化(己烷:etoac=92:8),得到int-141-6,作为黄色固体物(190mg,产率96%)。lcms:(m+1)m/z=449,451.int-141-7的合成将dmf(2ml)中的int-141-6(190mg,0.42mmol)、三乙基硼烷(1m的thf溶液,0.85ml)、k2co3(117mg,0.85mmol)和pd(dppf)cl2·ch2cl2(34mg,0.041mmol)的混合物用氮气脱气,并在70℃加热2h。将反应混合物冷却至rt,在硅藻土上过滤,并将产物在etoac和水之间进行分配。将有机相用盐水(3x20ml)洗涤,经na2so4干燥,并浓缩至干。将产物经柱色谱纯化,得到int-141-7,作为黄色固体物(45mg,产率27%)。lcms:(m+1)m/z=399.int-141-8的合成在rt,向化合物int-141-7(34mg,0.084mmol)中,加入10%aq.h2so4(0.28ml)。然后,将混合物在45℃搅拌2h。冷却至rt后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(2×10ml)。将合并的有机层经na2so4干燥,并浓缩至干,得到int-141-8,作为黄色固体物(29mg,定量产率)。lcms:(m+1)m/z=355.化合物141的合成将1,2-二氯乙烷(0.4ml)中的int-141-8(30mg,0.084mmol)、(r)-四氢-2h-吡喃-3-胺盐酸盐(15mg,0.11mmol)和dipea(20μl,0.11mmol)的混合物在室温下搅拌10min。向所述混合物中,加入nabh(oac)3(27mg,0.13mmol)和acoh(7μl,0.13mmol)。将得到的混合物在rt搅拌3h。将反应混合物直接经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物141,作为黄色固体物(32mg,产率87%)。lcms:(m+1)m/z=440.实施例142-146int-142-a的合成在0℃,向n-羟基乙脒(29g,0.39mol)和dipea(102ml,0.58mol)在1,4-二氧六环(200ml)中的混悬液中,滴加乙基丙二酰氯(50ml,0.39mol)。将混合物在rt搅拌20min,然后在100℃加热3h。冷却至rt后,将混合物减压浓缩。将残留物在盐水和醚之间分配。将有机层用饱和nahco3水溶液洗涤,经na2so4干燥,并浓缩。将残留物经短硅胶通路纯化,用20%etoac的己烷溶液洗脱,得到int-142-a,作为浅棕色液体(40.28g,产率61%)。int-142-9的合成在0℃,向噁二唑酯int-142-a(10.0g,58.9mmol)的thf(50ml)溶液中,加入2mnaoh水溶液(59ml)。然后,将反应物在rt搅拌1h。向其中,加入树脂amberliteir120(h+),以调节ph至ph4。滤掉树脂,并用h2o洗涤。将滤液减压浓缩。将残留物经p2o5真空干燥,得到int-142-9,并且不需纯化而用于下一步骤。int-142-2的合成向2-氨基-3-氟苯甲酸1(20.0g,129mmol)在ch2cl2(300ml)中的混悬液中,加入n-溴代琥珀酰亚胺(22.9g,129mmol)。将混合物在室温搅拌过夜。将产物经真空过滤而收集,得到2-氨基-5-溴-3-氟苯甲酸int-142-2,作为灰白色固体物(27g,产率90%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=232,234.int-142-3的合成将dmf(90ml)中的int-142-2(8.5g,36.3mmol)、n,o-二甲基羟基胺盐酸盐(6.4g,65.3mmol)、dipea(12.6ml)、edci(8.4g,43.6mmol)和hobt(6.7g,43.6mmol)的混合物在室温搅拌4h。然后,将反应物用100mletoac稀释,并依次用1mnaoh、1mhcl和盐水洗涤,得到int-142-3,作为棕色油状物(8.6g,产率85%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=277,279.int-142-4的合成在0℃,向int-142-3(8.6g,31mmol)的ch2cl2(100ml)溶液中,加入tea(5.2ml,37mmol),随后滴加三氟乙酸酐(5.6ml,40mmol)。将反应物在室温搅拌过夜。然后,加入饱和nahco3水溶液,并分离有机相,并用水洗涤。将有机层用na2so4干燥,并浓缩至干,得到int-142-4,作为黄色固体物(9.4g,80%产率),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=373,375int-142-5的合成向int-142-4(4.3g,11.5mmol)、cs2co3(11.0g,34.6mmol)和pd(dppf)cl2(170mg,0.23mmol)在thf(25ml)中的混悬液中,加入1mbet3的thf(34.6ml,34.6mmol)溶液,并将反应物在70℃搅拌1.5h。冷却至室温后,将粗品经硅藻土过滤并经柱色谱纯化(己烷:etoac),得到int-142-5,作为黄色固体物(2.5g,产率54%)。lcms:(m+1)m/z=323.int-142-6的合成在0℃,向int-142-5(2.0g,6.2mmol)的thf(40ml)溶液中,加入1.4mmemgbr的thf:甲苯溶液(22ml,31mmol),并将反应物在室温搅拌2.5h。然后,将反应物倒入冰中,用2mhcl酸化至ph=2,并用etoac萃取。将合并的有机层用na2so4干燥,并浓缩至干,得到int-142-6,作为黄色油状,没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=276.int-142-7的合成向int-142-6(2.0g,7.2mmol)的meoh(6ml)溶液中,加入2mnaoh水溶液(6ml)。然后,将反应物在90℃加热1.5h。加入水,并经过滤收集固体物,得到int-142-7,作为黄色固体物(1.2g,产率92%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=182.int-142-11的合成将酮int-142-7(250mg,1.38mmol)、2-(3-甲基-1,2,4-二唑-5-基)乙酸9(235mg,1.65mmol)和pocl3(2.5ml)的混合物在110℃搅拌1h。真空除去过量的pocl3。向残留物中,加入饱和nahco3水溶液,并将产物用etoac萃取。将合并的有机层用na2so4干燥,并浓缩至干。粗产物经柱色谱(己烷/etoac梯度)纯化,得到2-氯喹啉int-142-11,作为白色固体物(176mg,产率42%)。lcms:(m+1)m/z=306,308.int-142-12的合成向int-142-11(220mg,0.72mmol)的etoh(5ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(184μl,1.44mmol)和dipea(250μl,1.44mmol)。将混合物在110℃加热过夜。冷却至室温后,将混合物减压浓缩,并经柱色谱(己烷/etoac)纯化,得到缩酮int-142-12,作为黄色油状物(278mg,93%产率)。lcms:(m+1)m/z=413.int-142-13的合成向int-142-12(278mg,0.67mmol)的thf(1ml)溶液中,加入10%aq.h2so4(5ml)。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取,经na2so4干燥,并浓缩至干,得到酮int-142-13,作为微黄色油状物(224mg,产率90%),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=369.化合物142的合成将1,2-二氯乙烷(5.0ml)中的酮int-142-13(160mg,0.43mmol)、4-氨基四氢吡喃(66mg,0.65mmol)、nabh(oac)3(182mg,0.86mmol)和acoh(50μl,1.05mmol)的混合物在室温搅拌过夜。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh,95:5)纯化,得到化合物142,作为黄色固体物(178mg,产率91%)。lcms:(m+1)m/z=454.化合物143、化合物144、化合物145、化合物146的合成化合物编号143-146的合成是用与化合物142类似的方式,通过中间体13和适当胺的还原胺化进行。化合物143,浅黄色固体物,对于最后一步(还原胺化):产率89%。lcms:(m+1)m/z=424;化合物144,浅黄色固体物,对于最后一步(还原胺化):产率59%。lcms:(m+1)m/z=454;化合物145,浅黄色固体物,对于最后一步(还原胺化):产率59%。lcms:(m+1)m/z=440;化合物146,浅黄色固体物,对于最后一步(还原胺化):产率75%。lcms:(m+1)m/z=440。实施例147int-147-11的合成将二氧六环中的酮int-147-7(1.0当量)、2-氰基乙酸乙酯int-147-8(2.0当量)和nh4oac(5.0当量)的混合物在90℃加热8h。将混合物减压浓缩,并将固体物依次用水和etoac/己烷(1:9)(2x)洗涤。没有进一步纯化而使用浅黄色固体物(产率83-85%)。lcms:(m+1)m/z=231.将int-147-10(1.0当量)在pocl3中的混悬液在110℃搅拌1.5h。减压除去过量的pocl3。将粗品用冰淬灭,并将混合物在室温搅拌15min。将粗品经过滤收集,并用水(3x)洗涤。以定量产率得到产物int-147-11,作为浅黄色固体物。lcms:(m+1)m/z=249.int-147-12的合成将etoh中的int-147-11(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.5当量)和dipea(1.5当量)的混合物在110℃加热过夜。将混合物减压浓缩,并将产物经柱色谱纯化,利用己烷/etoac。以97-98%得到产物int-147-12,作为浅棕色固体物。lcms:(m+1)m/z=356.int-147-13的合成将无水异丙醇中的int-147-12(1.0当量)、nh2oh.hcl(5.0当量)和na2co3(5.0当量)的混合物在100℃加热过夜。将混合物冷却至室温并过滤。将有机相减压浓缩,并且没有进一步纯化而使用产物。lcms:(m+1)m/z=389.将二氧六环中的酰氨肟(1.0当量)、醋酸酐(1.2当量)和dipea(1.2当量)在室温搅拌40min,然后将混合物在100℃加热8h。将混合物减压浓缩,并将产物经柱色谱纯化,利用己烷/etoac,以64-66%产率(两步)得到浅棕色固体物。lcms:(m+1)m/z=413.向缩酮int-147-13(1.0当量)的thf(1ml)溶液中,加入10%aq.h2so4,并将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac(4x)萃取,经na2so4干燥,并浓缩至干,以定量产率得到浅棕色酮int-147-14,没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=369.化合物147的合成将1,2-二氯乙烷中的酮int-147-14(1.0当量)、4-氨基四氢吡喃(1.2当量)、nabh(oac)3(1.5当量)和acoh(1.5当量)的混合物在室温搅拌过夜。将混合物减压浓缩,并经制备型-tlc(ch2cl2:meoh,95:5)纯化,以90-92%产率得到化合物147,作为灰白色固体物。lcms:(m+1)m/z=454.实施例1483的合成将1,2-二氯乙烷(10ml)中的int-148-1(500mg,2.53mmol)、(r)-四氢-2h-吡喃-3-胺盐酸盐2(384mg,2.79mmol)和dipea(0.485ml,2.78mmol)的混合物在室温下搅拌10min。向所述混合物中,加入nabh(oac)3(803mg,3.79mmol)和acoh(0.22ml,3.79mmol)。将得到的混合物在rt搅拌过夜。将溶剂减压蒸发,将残留物在饱和na2co3水溶液和etoac之间分配。分离有机相,并将水层用etoac(2x20ml)萃取。将合并的有机相经na2so4干燥,并减压蒸发。将产物经柱色谱纯化(etoac:iproh=80:20),得到int-148-13,作为琥珀色油状物(705mg,产率98%)。lc-ms:(m+1)m/z=285.int-148-14的合成向int-148-13(703mg,2.47mmol)的ch2cl2(5ml)溶液中,滴加三氟乙酸(3.78ml,49mmol)。将反应混合物在rt搅拌1h,并将溶剂和过量的三氟乙酸减压蒸发,得到int-148-14,作为黄色油状物,没有进一步纯化,将其用于下一步骤(1.01g,定量产率)。lcms:(m+1)m/z=185.化合物148的合成向int-148-15(24.4mg,0.08mmol)的etoh(0.4ml)和2-丙醇(0.2ml)溶液中,加入int-148-14(r)-n-(四氢-2h-吡喃-3-基)哌啶-4-胺双三氟乙酸盐(33mg,0.08mmol)和dipea(55μl,0.32mmol)。将反应混合物用微波在140℃加热140min,然后冷却至rt,并用制备型-tlc纯化(ch2cl2:meoh=96:4)(2.6g,产率88%),得到化合物148,作为黄色固体物(10.0mg,27%产率)。lcms:(m+1)m/z=454.实施例149-152根据化合物147的方法,由酮int-147-14和相应的胺起始,得到化合物149-152。以88-90%产率得到化合物149,作为浅黄色固体物。lcms:(m+1)m/z=440.以89-90%产率得到化合物150,作为浅棕色固体物。lcms:(m+1)m/z=440.以82-83%产率得到化合物151,作为浅棕色固体物。lcms:(m+1)m/z=454.以71-75%产率得到化合物152,作为浅黄色固体物。lcms:(m+1)m/z=454.实施例153-155试剂和条件:i)int-153-15(1.0当量),int-153-8(2.0当量),nh4oac(5.0当量),90℃,8h,95-97%;ii)pocl3,110℃,1.5h,定量的;iii)int-153-17(1.0当量),1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.1当量),dipea(1.1当量),iproh,120℃,过夜,96-98%;iv)int-153-18(1.0当量),bet3(2.0当量),cs2co3(2.0当量),pd(dppf)cl2.ch2cl2(0.05当量),thf,70℃,1.5h,96-98%;v)int-153-19(1.0当量),nh2oh.hcl(5.0当量),na2co3(5.0当量),iproh,90℃,过夜;vi)(aco)2o(1.2当量),dipea(1.2当量),二氧六环,rt至90℃,8h,28-30%两步;vii)10%aq.h2so4,thf,45℃,2h,52-54%;viii)4-氨基四氢吡喃(1.2当量),nabh(oac)3(1.5当量),acoh(1.5当量),1,2-二氯乙烷,rt,过夜,78-80%.int-153-16的合成将二氧六环中的醛int-153-15(1.0当量)、2-氰基乙酸乙酯int-153-8(2.0当量)和nh4oac(5.0当量)的混合物在90℃搅拌8h。将混合物减压浓缩,并将固体物依次用水和etoac/己烷(1:9)(2x)洗涤。没有进一步纯化而使用黄色固体物int-153-16(产率95-97%)。lcms:(m+1)m/z=266,268.int-153-17的合成将int-153-16(1.0当量)在pocl3中的混悬液于110℃搅拌1.5h。减压除去过量的pocl3。将粗品用冰淬灭,并将混合物在室温搅拌15min。将粗品经过滤收集,并用水(3x)洗涤,以定量产率得到int-153-17,作为浅黄色固体物。lcms:(m+1)m/z=284,286.int-153-18的合成将iproh中的17(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.1当量)和dipea(1.1当量)的混合物在120℃加热过夜。将混合物减压浓缩,并将产物经柱色谱纯化,利用己烷/etoac。以96-98%得到产物int-153-18,作为浅棕色固体物。lcms:(m+1)m/z=392,394.int-153-19的合成向int-153-18(1.0当量)、cs2co3(2.0当量)和pd(dppf)cl2.ch2cl2(0.05当量)在thf中的混悬液中,加入1mbet3的thf(2.0当量)溶液,并将反应物在70℃加热1.5h。冷却至室温后,将粗品经硅藻土过滤,并经柱色谱纯化(己烷:etoac),得到int-153-19,作为黄色固体物(产率96-98%)。lcms:(m+1)m/z=342.int-153-20的合成将无水异丙醇中的int-153-19(1.0当量)、nh2oh.hcl(5.0当量)和na2co3(5.0当量)的混合物在90℃加热过夜。将混合物冷却至室温并过滤。将有机相减压浓缩,并且没有进一步纯化而使用产物。lcms:(m+1)m/z=375.将二氧六环中酰氨肟(1.0当量)、醋酸酐(1.2当量)和dipea(1.2当量)在室温搅拌40min,然后将混合物在90℃加热8h。将混合物减压浓缩,并将产物经柱色谱纯化,利用己烷/etoac,以28-30%产率得到浅黄色固体物int-153-20。lcms:(m+1)m/z=399.int-153-21的合成向缩酮int-153-20(1.0当量)的thf溶液中,加入10%aq.h2so4(12ml),并将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(4×),经na2so4干燥,并浓缩至干。将产物经柱色谱纯化,利用己烷/etoac,以52-54%产率得到浅黄色酮int-153-21。lcms:(m+1)m/z=355.化合物153的合成将1,2-二氯乙烷中的酮int-153-21(1.0当量)、4-氨基四氢吡喃(1.2当量)、nabh(oac)3(1.5当量)和acoh(1.5当量)混合物在室温搅拌过夜。将混合物减压浓缩,并用制备型-tlc纯化(ch2cl2:meoh,95:5),以78-80%产率得到化合物153,灰白色固体物。lcms:(m+1)m/z=440.化合物154和化合物155的合成以与酮int-153-21相同的方式,制备化合物154-155。以85-86%产率得到化合物155,作为浅黄色油状物。lcms:(m+1)m/z=426.以89-90%产率得到化合物154,作为浅黄色油状物。lcms:(m+1)m/z=426.实施例156-158int-156-28的合成向2-氨基-3,4-二氟苯甲酸27(1.0当量)在ch2cl2中的混悬液中,加入n-溴代琥珀酰亚胺(1.01当量),并将混合物在rt搅拌48h。将产物经过滤而收集,得到酸int-156-28,作为白色固体物(产率92-95%),没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=251,253.int-156-29的合成将dmf中的int-156-28(1.0当量)、n,o-二甲基羟基胺盐酸盐(1.2当量)、dipea(1.2当量)、edci(1.5当量)和hobt(1.5当量)的混合物在rt搅拌4h。将反应混合物用水稀释,并经过滤收集产物,作为白色固体物,没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=294,296。在0℃,向所述weinreb酰胺(1.0当量)和tea(1.2当量)的ch2cl2溶液中,滴加三氟乙酸酐(1.2当量)。将反应物在r.t.搅拌过夜。将混合物减压浓缩,并将粗品用水稀释。经过滤收集固体物,并且不需纯化,将其用于下一步骤。lcms:(m-1)m/z=388,390.int-156-30的合成将1met3b(3.0当量)的thf溶液加到int-156-29(1.0当量)、cs2co3(3.0当量)和pd(dppf)cl2(0.1当量)在thf中的混悬液中,并将反应物在50℃加热过夜。冷却至rt后,将粗品经硅藻土过滤,并将柱色谱纯化(己烷:etoac),以9-11%产率得到适当产物。lcms:(m+1)m/z=341.在0℃,向上述产物(1.0当量)的thf溶液中,加入memgbr(1.4m)的thf:甲苯(4.0当量)溶液,并将反应物在室温搅拌2.5h。将反应物倒入冰中,用2mhcl酸化至ph2,并将产物用etoac(2x)萃取。将合并的有机层用na2so4干燥,并浓缩,得到相应的酮,没有进一步纯化,将其用于下一步骤。lcms:(m-1)m/z=294.向酮(1.0当量)的meoh溶液中,加入2mnaoh水溶液(2.0当量),并将反应物在90℃加热1.5h。将反应混合物浓缩,并用1mhcl酸化。将产物用etoac萃取(3x),并将有机相减压浓缩。将粗品经柱色谱利用己烷:etoac纯化,得到int-156-30,作为浅黄色固体物(两步产率50-54%)。lcms:(m+1)m/z=200.int-156-31的合成将int-156-30(1.0当量)、2-(3-甲基-1,2,4-二唑-5-基)乙酸2(1.0当量)和pocl3(2.5ml)的混合物在80℃搅拌1h。减压除去过量的pocl3。向残留物中加入冰/水,并将固体物经过滤收集。没有进一步纯化而使用粗品。lcms:(m+1)m/z=324,326.int-156-32的合成将int-156-31(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(1.2当量)和dipea(1.2当量)在iproh中的混悬液于125℃加热过夜。冷却至室温后,将混合物减压浓缩,并经柱色谱纯化(己烷:etoac),得到缩酮int-156-32,作为黄色固体物(两步产率47-49%).lcms:(m+1)m/z=431.int-156-33的合成将缩酮int-156-32(1.0当量)在10%aq.h2so4中的混悬液在45℃搅拌2h。冷却至rt后,将混合物用na2co3中和,并用etoac萃取(3×)产物。将有机相经na2so4干燥并浓缩,得到酮int-156-33,作为微黄色固体物,没有进一步纯化而将其用于下一步骤。lcms:(m+1)m/z=387.化合物156、化合物157和化合物158的合成将1,2-二氯乙烷中的酮int-156-33(1.0当量)、适当的胺(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在rt搅拌过夜。将混合物减压浓缩,并经hplc纯化,得到目标化合物。得到化合物156,作为浅棕色固体物,产率96-97%(最后一步)。lcms:(m+1)m/z=472.得到化合物157,作为浅棕色固体物,产率79-81%(最后一步)。lcms:(m+1)m/z=472.得到化合物158,作为浅黄色油状物,产率94-96%(最后一步)。lcms:(m+1)m/z=458.实施例159-160int-159-2的合成在-78℃,向int-159-1(1.23g,5mmol)的thf(25ml)溶液中,加入n-buli溶液(2.5m的己烷,6ml,15mmol),并将混合物在-78℃搅拌1h。在-78℃,加入乙醛(1.1ml,20mmol),并将混合物在-78℃搅拌0.5小时。将反应用饱和nh4cl水溶液淬灭,并用etoac(3x)萃取产物。将有机层经na2so4干燥,并浓缩至干,得到int-159-2,作为黄色液体,没有进一步纯化,将其用于下一步。lcms:(m+1)m/z=290.int-159-3的合成在室温,向int-159-2的ch2cl2(30ml)溶液中,加入dmp(3.18g,7.5mmol)和nahco3(0.84g,10mmol),并将混合物搅拌2小时。向混合物中加入己烷(30ml),并将合并的混悬液经短硅胶柱过滤。用己烷/etoac(4:1,50ml×3)洗脱柱子。将合并的有机溶液浓缩,得到int-159-3,作为黄色固体物(1.34g,两步产率93%),没有进一步纯化,将其用于下一步骤。lcms:(m+23)m/z=312.int-159-7的合成向int-159-3(288mg,1mmol)的thf(4ml)溶液中,加入pd(oac)2(6.7mg,0.03mmol)、xphos(28.6mg,0.06mmol)、et3b(1.0m的thf溶液,3ml,3mmol)和cs2co3(975mg,3mmol)。将反应混合物在80℃搅拌过夜。冷却至室温后,将粗品经硅藻土过滤,并经柱色谱纯化(己烷:etoac),得到int-159-7,作为黄色液体(176mg,产率51%).lcms:(m+23)m/z=304.int-159-8的合成将ch2cl2(4ml)中的int-159-7(150mg,0.53mmol)和tfa(1ml)的混合物在室温搅拌4小时。将反应物用饱和na2co3水溶液淬灭,并用ch2cl2萃取产物。将合并的有机层经na2so4干燥,并浓缩,得到int-159-8,作为黄色液体,没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=182.int-159-9的合成将int-159-8、2-(3-甲基-1,2,4-二唑-5-基)乙酸(110mg,0.77mmol)和pocl3(1.5ml)的混合物在110℃搅拌过夜。真空除去过量的pocl3。向残留物中加入饱和nahco3水溶液,用etoac萃取产物。将合并的有机层用na2so4干燥,并浓缩至干。粗产物经柱色谱(己烷/etoac)纯化,得到int-159-9,作为黄色固体物(60mg,两步产率39%)。lcms:(m+1)m/z=306.int-159-10的合成向int-159-9(50mg,0.16mmol)的etoh(1.6ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(41μl,0.32mmol)和dipea(56μl,0.32mmol)。将反应混合物在110℃加热过夜。冷却至室温后,将混合物减压浓缩并经柱色谱纯化(己烷/etoac),得到缩酮int-159-10,作为黄色固体物(60mg,产率91%).lcms:(m+1)m/z=413.int-159-11的合成向int-159-10(1.0当量)的丙酮溶液中,加入2mhcl水溶液,并将混合物在rt搅拌5h。将反应物用etoac稀释,并依次用na2co3水溶液和盐水洗涤,以91-93%产率得到int-159-11,作为浅棕色固体物,没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=369.化合物159的合成将1,2-二氯乙烷中的酮int-159-11(1.0当量)、4-氨基四氢吡喃(1.2当量)、nabh(oac)3(1.5当量)和acoh(1.5当量)的混合物在室温搅拌过夜。将混合物减压浓缩,并经hplc纯化,以91-93%产率得到化合物159,作为灰白色固体物。lcms:(m+1)m/z=454.化合物160的合成用与化合物159相同的方式,利用中间体酮int-159-11和相应的胺,得到化合物160,作为浅黄色油状物;81%产率。lcms:(m+1)m/z=454.实施例161-165int-161-2的合成向苯胺int-161-1(1.0当量)在ch2cl2(100ml)的混悬液中,加入nbs(1.05当量),并将反应混合物在室温搅拌过夜。将混合物在饱和nahco3水溶液和ch2cl2之间分配。分离有机层,经na2so4干燥并浓缩。经柱色谱纯化(己烷/etoac),得到int-161-2,作为黄色固体物(产率57-59%)。lcms:(m+1)m/z=223,225.int-161-3的合成在-78℃,向int-161-2(1.0当量)的thf溶液中,滴加2.0mnbuli(2.2当量)的己烷溶液。在-78℃搅拌1h后,在相同温度下,加入dmf(1.4当量)。将温度升至0℃,并将混合物在0℃搅拌2h。将混合物用饱和nh4cl水溶液淬灭,并用etoac(2x)萃取产物。将有机相用盐水(3x)洗涤,经硫酸钠干燥,并减压浓缩。经柱色谱纯化(己烷/etoac梯度),得到int-161-3,作为黄色固体物(53-55%产率)。lcms:(m+1)m/z=174.int-161-5的合成将pocl3中int-161-3(1.0当量)和4(1.2当量)的混合物在110℃搅拌1.5h。减压浓缩过量的pocl3。将粗品用冰淬灭,并在室温搅拌15min。经真空过滤收集固体物,并用水洗涤(2x)。将固体物用短硅胶柱利用己烷/etoac纯化,以17-19%产率得到化合物int-161-5,作为黄色固体物。lcms:(m+1)m/z=397,399.int-161-6的合成将int-161-5(1.0当量)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(2.0当量)和dipea(2.0当量)的iproh溶液在110℃加热过夜。冷却至室温后,将混合物浓缩并经柱色谱纯化(己烷/etoac),得到int-161-6,作为黄色固体物(产率82-84%)。lcms:(m+1)m/z=405.int-161-7的合成在室温,向int-161-6(1.0当量)的thf溶液中,加入10%aq.h2so4。将混合物在45℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(×3)。将合并的有机层经na2so4干燥,并减压浓缩。将残留物经柱色谱纯化(己烷/etoac梯度),得到int-161-7,作为浅黄色固体物(产率63-65%)。lcms:(m+1)m/z=361.化合物161的合成将1,2-二氯乙烷中的int-161-7(1.0当量)、胺(2.0当量)、nabh(oac)3(2.0当量)和acoh(2.0当量)的混合物在室温搅拌过夜。经硅藻土过滤后,将滤液减压浓缩。残留物经制备型-tlc利用ch2cl2/meoh纯化。以71-73%产率得到化合物161,作为浅黄色固体物。lcms:(m+1)m/z=446.化合物162、化合物163、化合物164、化合物165的合成根据化合物160的方法,由酮int-161-14和相应的胺开始,得到标题化合物。以65-67%产率得到化合物162,作为浅黄色固体物。lcms:(m+1)m/z=446.以66-68%产率得到化合物163,作为浅黄色固体物。lcms:(m+1)m/z=446.以59-61%产率得到化合物164,作为浅黄色固体物。lcms:(m+1)m/z=432.以72-74%产率得到化合物165,作为浅黄色固体物。lcms:(m+1)m/z=432.实施例166利用上文化合物33-35的合成中所述的酮int-33-14k,制备化合物166。将1,2-二氯乙烷(0.1ml)中的酮int-33-14k(15mg,0.040mmol)、氧杂环丁-3-基甲胺(5.3mg,0.060mmol)和acoh(5μl,0.081mmol)的混合物在室温搅拌30分钟,然后加入nabh(oac)3(17.2mg,0.081mmol)。将混合物在室温搅拌过夜。将混合物减压浓缩并经制备型-tlc纯化(ch2cl2:meoh,95:5),得到化合物166,作为黄色固体物(2.3mg,产率12.9%)。lcms:(m+1)m/z=442.实施例167-169如上文对化合物40-59和化合物134-137所公开,利用具有适当立体化学的适当的胺,以及上文化合物36-39制备中所述的int-36-14h,得到化合物167-169实施例170-171int-170-1a的合成从上文化合物91的合成中所述的int-91-22开始,并根据上文化合物33-35的合成中所述的int-33-14j所公开,得到中间体int-170-1a。1b的合成根据上文化合物33-35的合成中所述的int-33-14k所公开,得到中间体int-170-1b。化合物170和化合物171的合成根据上文化合物33-35的合成中所公开,由int-170-1b和适当的胺,得到化合物170和化合物171。实施例172反应试剂和条件(int-172-4andint-172-13为例):i)int-172-1(1.0eq.),胺(int-172-2,1.2eq.),nabh(oac)3(1.5eq.),acoh(1.5eq.),1,2-二氯乙烷,r.t.,18h,87-97%;ii)int-172-3(1.0eq.),tfa(20eq.),ch2cl2,r.t.,40min;iii)int-172-1(1.0eq.),int-172-11(2.0eq.),dipea(3.0eq.),ch3cn,95℃,48h,68-95%.2-甲基-4-((四氢-2h-吡喃-4-基)氨基)哌啶-1-甲酸(2r,4r)-叔丁酯int-172-3的合成向胺int-172-1(40mg,0.187mmol)和酮int-172-2(20.8μl,0.22mmol)的dce(300μl)溶液中,加入nabh(oac)3(59mg,0.28mmol)和acoh(15.5μl,0.28mmol)。将得到的混合物在室温搅拌过夜。将混合物用1mnaoh淬灭并用etoac萃取(4x)。将有机层经na2so4干燥,并浓缩。经柱色谱纯化(ch2cl2/meoh梯度)得到化合物int-172-3,作为浅灰色固体物(54mg,产率97%)。lcms:(m+1)m/z=299.(2r,4r)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-172-4的合成将ch2cl2(2ml)中的int-172-3(54mg,0.18mmol)和tfa(277μl,3.61mmol)的混合物在室温搅拌40min。将混合物减压浓缩。将粗品再溶解于etoac中,并缓慢滴加几滴浓的naoh水溶液。加入无水mgso4,除去h2o,并滤出固体物,并用etoac(3x)洗涤。将滤液浓缩,并且没有进一步纯化而使用产物(定量产率)。得到int-172-4,作为浅黄色油状物。lcms:(m+1)m/z=199.(2r,4s)-2-甲基-4-((四氢-2h-吡喃-4-基)氨基)哌啶-1-甲酸叔丁酯int-172-6的合成向胺int-172-5(40mg,0.187mmol)和酮int-172-2(20.8μl,0.22mmol)的dce(300μl)溶液中,加入nabh(oac)3(59mg,0.28mmol)和acoh(15.5μl,0.28mmol)。将得到的混合物在室温搅拌过夜。将混合物用1mnaoh淬灭并用etoac萃取(4x)。将有机层经na2so4干燥,并浓缩。经柱色谱纯化(ch2cl2/meoh梯度)得到化合物int-172-6,作为浅灰色固体物(53mg,产率97%)。lcms:(m+1)m/z=299.(2r,4s)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-172-7的合成将ch2cl2(2ml)中的int-172-6(53mg,0.18mmol)和tfa(277μl,3.61mmol)的混合物在室温搅拌40min。将混合物减压浓缩。将粗品再溶解于etoac中,并缓慢滴加几滴浓的naoh水溶液。加入无水mgso4,除去h2o,并滤出固体物,并用etoac(3x)洗涤。将滤液浓缩,并且没有进一步纯化而使用产物(定量产率)。得到int-172-7,作为浅黄色油状物。lcms:(m+1)m/z=199.(2s,4s)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-172-10的合成用相似方式,以94%产率(通过两步)合成化合物(2s,4s)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-172-10,并作为浅黄色油状物而得到。lcms:(m+1)m/z=199.(2r4r)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-172-13的合成将乙腈(10ml)中的int-172-1(1.0g,4.6mmol)、int-172-11(3.3g,14mmol)和dipea(2.44ml,14mmol)的混合物在95℃加热48h。将混合物减压浓缩,并经柱色谱纯化,利用ch2cl2/meoh作为流动相,得到1.0g浅灰色固体物(76%产率)。lcms:(m+1)m/z=285.将ch2cl2中的int-172-12(1g,3.51mmol)和tfa(5.38ml,70.3mmol)的混合物在室温搅拌40min。将混合物减压浓缩。将粗品再溶解于etoac中,并缓慢滴加几滴浓的naoh水溶液。加入无水mgso4,除去h2o,并滤出固体物,并用etoac(3x)洗涤。将滤液浓缩,并且没有进一步纯化而使用产物(640mg,定量产率)。lcms:(m+1)m/z=185.(2r4r)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺int-172-16的合成将乙腈(10ml)中的int-172-1(1.0g,4.6mmol)、int-172-14(3.3g,14mmol)和dipea(2.44ml,14mmol)的混合物在95℃加热48h。将混合物减压浓缩,并经柱色谱纯化,利用ch2cl2/meoh作为流动相。得到1.26g浅灰色固体物(95%产率)。lcms:(m+1)m/z=285.将ch2cl2(20ml)中的int-172-15(1260mg,4.43mmol)和tfa(6.78ml,88.6mmol)的混合物在室温搅拌40min。将混合物减压浓缩。将粗品再溶解于etoac中,并缓慢滴加几滴浓的naoh水溶液。加入无水mgso4,除去h2o,并滤出固体物,并用etoac(3x)洗涤。将滤液浓缩,并且没有进一步纯化而使用产物(810mg,定量产率)。lcms:(m+1)m/z=185.(2r4s)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-172-18的合成用相似方式,合成化合物(2r,4s)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-172-18。以46%产率(两步)得到产物,作为浅黄色油状物。lcms:(m+1)m/z=185.(2s,4s)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-172-20的合成用相似方式,合成化合物(2s,4s)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-172-20。以33%产率(两步)得到产物,作为浅黄色油状物。lcms:(m+1)m/z=185.反应试剂和条件(int-172-26为例):i)int-172-21(1.0eq.),nis(1.05eq.),acoh,r.t.,67-74%;ii)int-172-22(1.0eq.),int-172-23(6.0eq.),k2co3(1.2eq.),dppp(0.05eq.),pd(oac)2(0.01eq),甲苯/h2o(1∶9)90℃,24-40h,10-37%;iii)int-172-24(1.0eq.),int-172-25(1.2eq.),pocl3,80℃,1h,17-95%;iv)int-172-42(1.0eq),nbs(1.05eq),ch2cl2,r.t.,18h,56%;v)int-172-38(1.0eq.),二苯甲酮亚胺(1.2eq.),pd2(dba)3(0.025eq.),xantphos(0.1eq.),naotbu(1.2eq.),1,4-二氧六环100℃,18h;随后hcl(1maq.),thf,r.t.,1h,30%.5-(2,8-二氯-4,6-二甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-172-26的合成2-氯-6-碘-4-甲基苯胺int-172-22的合成向int-172-21(5.0g,35.3mmol)的乙酸(50ml)混合物中,一次性加入nis(8.34g,37.1mmol),并将混合物在室温搅拌1h。将混合物用etoac(500ml)稀释,并用盐水(3x)和饱和nahco3水溶液(2x)洗涤。将有机相用na2so4干燥,并减压浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~30%etoac的己烷溶液)作为流动相。以74%产率得到产物,作为淡红色固体物(7g)。lcms:(m+1)m/z=267.1-(2-氨基-3-氯-5-甲基苯基)乙酮int-172-24的合成将60mlh2o/甲苯(9:1)中的int-172-22(7g,26.17mmol)、23(14.09ml,157mmol)、k2co3(4.34g,31.4mmol)、dppp(521mg,1.3mmol)和pd(oac)2(59mg,0.26mmol)的混合物在90℃加热24h。将混合物冷却至室温后,缓慢加入浓hcl(15ml),并将混合物在室温搅拌1h。将产物用etoac(3x)萃取。将有机相经无水na2so4干燥,并减压浓缩。将产物经柱色谱利用己烷/etoac(0~20%etoac的己烷溶液)纯化,并以27%产率得到产物,作为浅黄色固体物(1.3g)。lcms:(m+1)m/z=184.5-(2,8-二氯-4,6-二甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-172-26的合成将pocl3(5ml)中的int-172-24(1.3g,7.08mmol)和25(1.2g,8.5mmol)的混合物在80℃搅拌1h。将混合物减压浓缩并用冰/h2o淬灭。将混合物在室温搅拌30min,并过滤产物。将固体经柱色谱纯化,利用己烷/etoac(0~30%etoac的己烷溶液),并以19%产率得到产物,作为浅黄色固体物(430mg)。lcms:(m+1)m/z=308.5-(2,6-二氯-8-氟-4-甲基喹啉-3-基)-3-甲基-1,24-二唑29的合成1-(2-氨基-5-氯-3-氟苯基)乙烷-1-酮int-172-28的合成将60mlh2o/甲苯(9:1)中的int-172-27(5g,18.4mmol)、23(9.9ml,110.5mmol)、k2co3(3.05g,22.1mmol)、dppp(366mg,0.92mmol)和pd(oac)2(41mg,0.18mmol)的混合物在90℃加热24h。将混合物冷却至室温后,缓慢加入浓hcl(14ml),并将混合物在室温搅拌1h。将产物用etoac(3x)萃取。将有机相经无水na2so4干燥,并减压浓缩。将产物经柱色谱利用己烷/etoac(0~20%etoac的己烷溶液)纯化,并以37%产率得到产物,作为浅棕色固体物(1.3g)。lcms:(m+1)m/z=188.5-(2,6-二氯-8-氟-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-172-29的合成将pocl3(5ml)中的int-172-28(1.25g,6.66mmol)和25(1.04g,7.33mmol)的混合物在80℃搅拌1h。将混合物减压浓缩并用冰/h2o淬灭。将混合物在室温搅拌30min,并过滤产物。将固体经柱色谱利用己烷/etoac(0~30%etoac的己烷溶液)纯化,并以17%产率得到产物,作为浅棕色固体物(350mg)。lcms:(m+1)m/z=312.5-(2-氯-8-氟-4-甲基-6-(三氟甲氧基)喹啉-3-基)-3-甲基-1,2,4-二唑int-172-33的合成2-氟-6-碘-4-(三氟甲氧基)苯胺int-172-31的合成将1,4-二氧六环(100ml)中的1-溴-2-氟-4-(三氟甲氧基)苯int-172-38(12.8g,49.4mmol)、二苯酮亚胺(10ml,59.3mmol)、pd2(dba)3(1.1g,1.23mmol)、xantphos(2.8g,4.9mmol)和naotbu(5.7g,59.3mmol)的混合物搅拌,并在100℃下加热过夜。冷却至室温后,将混合物浓缩。然后,将残留物在etoac和h2o之间分配,并分离有机层,经na2so4干燥,并浓缩。经柱色谱纯化,利用己烷/etoac(0~5%etoac的己烷溶液)。将产物溶解于thf(100ml)中,并加入1mhcl水溶液(50ml)。在室温搅拌1h后,将混合物在etoac和h2o之间进行分配。分离有机层,并用盐水洗涤,na2so4干燥并浓缩。经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)。以30%产率得到产物30,作为浅棕色油状物(5.4mg)。lcms:(m+1)m/z=196.向int-172-30(310mg,1.58mmol)的乙酸(5ml)混合物中,一次性加入nis(373mg,1.66mmol),并将混合物在室温搅拌2.5h。将混合物用etoac稀释,并用盐水(3x)和饱和na2co3(2x)水溶液洗涤。将有机层经na2so4干燥,并真空浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以67%产率得到产物,作为红色油状物(340mg)。1-(2-氨基-3-氟-5-(三氟甲氧基)苯基)乙酮int-172-32的合成将1,4-二氧六环/h2o(2ml,9:1v/v)中的int-172-31(340mg,1.06mmol)、int-172-23(0.5ml,5.30mmol)、k2co3(176mg,1.27mmol)、dppp(22mg,0.053mmol)和pd(oac)2(2.4mg,0.0106mmol)的混合物在90℃加热过夜。将混合物冷却至室温后,缓慢加入浓hcl(1ml),并将混合物在室温搅拌1h。将产物用etoac(3x)萃取。将合并的有机层经无水na2so4干燥,并真空浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以51%产率得到产物,作为浅黄色固体物(128mg)。lcms:(m+1)m/z=238.5-(2-氯-8-氟-4-甲基-6-(三氟甲氧基)喹啉-3-基)-3-甲基-1,2,4-二唑int-172-33的合成将pocl3(2ml)中的int-172-32(128mg,0.54mmol)和int-172-25(92mg,0.65mmol)的混合物在100℃搅拌1h。冷却至室温后,减压除去过量的pocl3。在0℃,向残留物中加入水,并将混合物在0℃搅拌10min。将沉淀的粗氯喹啉int-172-33过滤,用h2o洗涤,并减压干燥。没有进一步纯化而使用残留物,作为黑褐色固体物(185mg,95%产率)。lcms:(m+1)m/z=362.2-氯-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-8-腈int-172-37的合成2-氨基-3-碘-5-甲基苄腈int-172-35的合成向int-172-34(5.0g,37.8mmol)的乙酸(50ml)混合物中,一次性加入nis(8.9g,39.7mmol),并将混合物在室温搅拌1h。将混合物用etoac(500ml)稀释,并用盐水(3x)和饱和nahco3水溶液(2x)洗涤。将有机相经na2co3干燥,并减压浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~30%etoac的己烷溶液)作为流动相。以71%产率得到35,作为浅棕色固体物(6.9g)。lcms:(m+1)m/z=258.3-乙酰基-2-氨基-5-甲基苄腈int-172-36的合成将40mlh2o/甲苯(9:1)中的int-172-35(2g,7.75mmol)、int-172-23(4.17ml,46.5mmol)、k2co3(1.28g,9.3mmol)、dppp(154mg,0.39mmol)和pd(oac)2(17mg,0.08mmol)的混合物在90℃加热24h。将混合物冷却至室温后,缓慢加入浓hcl(8ml),并将混合物在室温搅拌1h。将产物用etoac(3x)萃取。将有机相经无水na2so4干燥,并减压浓缩。将产物经柱色谱利用己烷/etoac(0~30%etoac的己烷溶液)纯化,并以15%产率得到产物,作为浅黄色固体物(203mg)。lcms:(m+1)m/z=175.2-氯-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-8-腈int-172-37的合成将pocl3(4ml)中的int-172-36(408mg,2.3mmol)和int-172-25(400mg,2.8mmol)的混合物在80℃搅拌1h。将混合物减压浓缩并用冰/h2o淬灭。将混合物在室温搅拌30min,并过滤产物。将固体经柱色谱利用己烷/etoac(0~40%etoac的己烷溶液)纯化,并以28%产率得到产物,作为浅黄色固体物(190mg)。lcms:(m+1)m/z=299.5-(2-氯-6,8-二氟-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-172-41的合成1-(2-氨基-3,5-二氟苯基)乙酮int-172-40的合成将1,4-二氧六环/h2o(25ml,9:1,v/v)中的int-172-39(3.02g,11.84mmol)、23(5.3ml,59.20mmol)、k2co3(2.0g,14.20mmol)、dppp(0.24g,0.59mmol)和pd(oac)2(27mg,0.12mmol)的混合物在90℃加热24h。将混合物冷却至室温后,缓慢加入浓hcl(6ml),并将混合物在室温搅拌1h。将产物用etoac(3x)萃取。将合并的有机层经na2so4干燥,并真空浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以28%产率得到产物,作为黄色固体物(0.56g)。lcms:(m+1)m/z=172.5-(2-氯-68-二氟-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑int-172-41的合成将pocl3(10ml)中的int-172-40(0.56g,3.28mmol)和int-172-25(0.56g,3.94mmol)的混合物在100℃搅拌1h。冷却至室温后,真空除去过量的pocl3。在0℃,向残留物中加入水,并将混合物在0℃搅拌10min。将沉淀的粗氯喹啉int-172-41过滤,用h2o洗涤,并减压干燥。将固体物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以25%产率得到产物,作为浅棕色固体物(0.25g)。lcms:(m+1)m/z=296.5-(2-氯-8-氟-4-甲基-6-(三氟甲基)喹啉-3-基)-3-甲基-1,2,4-二唑int-172-45的合成2-溴-6-氟-4-(三氟甲基)苯胺int-172-43的合成向int-172-42(8.5g,47.45mmol)在ch2cl2(100ml)的混悬液中,一次性加入nbs(8.9g,49.83mmol),并将混合物在室温搅拌过夜。真空浓缩后,将混合物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以56%产率得到为红色油状物的产物(6.8g)。1-(2-氨基-3-氟-5-(三氟甲基)苯基)乙酮int-172-44的合成将1,4-二氧六环/h2o(60ml,9:1,v/v)中的int-172-43(6.8g,26.35mmol)、int-172-23(12ml,131.75mmol)、k2co3(4.4g,31.62mmol)、dppp(0.55g,1.32mmol)和pd(oac)2(59mg,0.26mmol)的混合物在95℃加热40h。将混合物冷却至室温后,缓慢加入浓hcl(14ml),并将混合物在室温搅拌1h。将产物用etoac(3x)萃取。将合并的有机层经na2so4干燥,并真空浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以10%产率得到产物,作为黄色固体物(0.58g)。lcms:(m+1)m/z=222.5-(2-氯-8-氟-4-甲基-6-(三氟甲基)喹啉-3-基)-3-甲基-1,2,4-二唑int-172-45的合成将pocl3(5ml)中的int-172-44(0.58g,2.64mmol)和int-172-25(0.45g,3.17mmol)的混合物在100℃搅拌1h。冷却至室温后,真空除去过量的pocl3。在0℃,向残留物中加入h2o,并将混合物在0℃搅拌10min。将沉淀的粗氯喹啉int-172-45过滤,用h2o洗涤,并减压干燥。将固体物经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以26%产率得到产物,作为浅棕色固体物(0.24g)。lcms:(m+1)m/z=346,348.反应试剂和条件:i)int-172-46(1.0eq.),胺(1.5eq.),kf(2.5eq.),dmf,125℃,18h,10-21%.(2r,4s)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺的合成(化合物172)将dmf(150μl)中的int-172-46(10mg,0.033mmol)、7(9.7mg,0.049mmol)和kf(4.7mg,0.08mmol)的混合物在125℃搅拌过夜。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc利用ch2cl2/meoh(95∶5)纯化。以21%产率得到化合物172,作为浅棕色油状物(3.2mg)。lcms:(m+1)m/z=468.以10%产率得到(2s,4s)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺化合物173,作为浅黄色油状物(4.1mg)。lcms:(m+1)m/z=468.实施例174-176反应试剂和条件:i)int-174-26(1.0eq.),胺(1.5eq.),kf(2.5eq.),dmso,125℃,14h,17-20%.(2r,4s)-1-(8-氯-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物174)的合成将dmso(200μl)中的int-174-26(20mg,0.062mmol)、int-174-4(19mg,0.097mmol)和kf(9.4mg,0.163mmol)的混合物在125℃搅拌14h。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc利用ch2cl2/meoh(95∶5)纯化。以17%产率得到化合物174,作为浅黄色油状物(5.3mg)。lcms:(m+1)m/z=470.以20%产率,得到(2r,4s)-1-(8-氯-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物175),作为浅黄色油状物(5.8mg)。lcms:(m+1)m/z=456.以20%产率,得到(2r,4s)-1-(8-氯-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺化合物176,作为浅黄色油状物(5.8mg)。lcms:(m+1)m/z=456.实施例177-179试剂和条件:i)int-177-29(1.0eq.),胺(2.0eq.),kf(2.5eq.),dmso,125℃,14h,14-33%.(2r,4r)-1-(6-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物177)的合成将dmso(200μl)中的int-174-29(20mg,0.064mmol)、int-177-4(25mg,0.127mmol)和kf(9.3mg,0.159mmol)的混合物在125℃搅拌过夜。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc利用ch2cl2/meoh(95∶5)纯化。以14%产率得到化合物177,作为浅棕色油状物(4.3mg)。lcms:(m+1)m/z=474.以33%产率,得到(2r,4r)-1-(6-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物178),作为浅棕色油状物(9.7mg)。lcms:(m+1)m/z=460.以30%产率,得到(2r,4r)-1-(6-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物179),作为浅棕色油状物(8.7mg)。lcms:(m+1)m/z=460.实施例180反应试剂和条件:i)int-180-33(1.0eq.),int-180-13(1.5eq.),kf(2.0eq.),dipea(3.0eq.),dmso,140℃,18h,10%.(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-6-(三氟甲氧基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物180)的合成将dmso(1ml)中的int-180-33(15mg,0.041mmol)、int-180-13(11mg,0.062mmol)、kf(4.7mg,0.082mmol)和dipea(21μl,0.123mmol)的混合物在140℃搅拌过夜。将混合物冷却至室温,用ch2cl2稀释,并用盐水洗涤。将有机相经na2so4干燥,并真空浓缩。将粗品经prep-tlc,利用ch2cl2/meoh(96∶4),然后用etoac∶iproh(95∶5)纯化。以10%产率得到化合物180,作为黄色固体物(2.0mg)。lcms:(m+1)m/z=510.实施例181-183反应试剂和条件:i)int-181-37(1.0eq.),胺(2.0eq.),kf(2.5eq.),dmso,125℃,14h,14-17%.4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-((四氢-2h-吡喃-4-基)氨基)哌啶-1-基)喹啉-8-腈(化合物181)的合成将dmso(200μl)中的int-181-37(20mg,0.066mmol)、int-181-4(18.4mg,0.1mmol)和kf(9.6mg,0.165mmol)的混合物在125℃加热14h。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc利用ch2cl2/meoh(95∶5)纯化。以14%产率得到化合物181,作为浅黄色油状物(4.3mg)。lcms:(m+1)m/z=461.以17%产率,得到4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-(((s)-四氢呋喃-3-基)氨基)哌啶-1-基)喹啉-8-腈(化合物182),作为浅棕色油状物(5.1mg)。lcms:(m+1)m/z=447.以15%产率,得到4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-(((r)-四氢呋喃-3-基)氨基)哌啶-1-基)喹啉-8-腈(化合物183),作为浅棕色油状物(4.3mg)。lcms:(m+1)m/z=447.实施例184-186反应试剂和条件:i)int-184-41(1.0eq.),胺(1.5eq.),kf(2.0eq.),dipea(3.0eq.),dmso,125℃,18h,6-22%.(2r,4r)-1-(6,8-二氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物184)的合成将dmso(1ml)中的int-184-41(25mg,0.084mmol)、int-184-16(23mg,0.126mmol)、kf(10mg,0.168mmol)和dipea(44μl,0.252mmol)的混合物在125℃加热过夜。将混合物冷却至室温,用ch2cl2稀释,并用盐水洗涤。将有机相经na2so4干燥,并真空浓缩。将粗品经prep-tlc,利用etoac∶iproh(97∶3),然后用ch2cl2/meoh(95∶5)纯化。以6%产率得到化合物184,作为浅黄色固体物(2.1mg)。lcms:(m+1)m/z=444.以12%产率,得到(2r,4r)-1-(6,8-二氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物185),作为浅黄色固体物(4.5mg)。lcms:(m+1)m/z=444.以22%产率,得到(2r,4r)-1-(6,8-二氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物186),作为浅黄色固体物(7.5mg)。lcms:(m+1)m/z=458.实施例187-189反应试剂和条件i)int-187-45(1.0eq.),胺(1.5eq.),kf(2.0eq.),dipea(3.0eq.),dmso,125℃,18h,14-33%.(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-6-(三氟甲基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物187)的合成将dmso(1ml)中的int-187-45(25mg,0.072mmol)、int-187-16(20mg,0.108mmol)、kf(8.4mg,0.144mmol)和dipea(38μl,0.216mmol)的混合物在125℃加热过夜。将混合物冷却至室温,用ch2cl2稀释,并用盐水洗涤。将有机相经na2so4干燥,并真空浓缩。将粗品经prep-tlc利用etoac∶iproh(98∶2),然后用ch2cl2/meoh(97∶3)纯化。以14%产率得到化合物187,作为浅橙色固体物(4.8mg)。lcms:(m+1)m/z=494.以31%产率,得到(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-6-(三氟甲基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物188),作为浅橙色固体物(11.1mg)。lcms:(m+1)m/z=494.以33%产率,得到(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-6-(三氟甲基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物189),作为浅橙色固体物(12.2mg)。lcms:(m+1)m/z=508.实施例190-192反应试剂和条件i)int-190-47(1.0eq.),胺(2.0eq.),kf(2.5eq.),dmso,125℃,14h,23-24%.(2r,4r)-1-(8-氯-6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氧-2h-吡喃-4-基)哌啶-4-胺(化合物190)的合成将dmso(200μl)中的int-190-47(20mg,0.062mmol)、int-190-4(15mg,0.074mmol)和kf(9mg,0.155mmol)的混合物在125℃加热过夜。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc利用ch2cl2/meoh(95∶5)纯化。以23%产率得到化合物190,作为作为浅橙色油状物(7mg)。lcms:(m+1)m/z=484.以23%产率,得到(2r,4r)-1-(8-氯-6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物191),作为浅棕色油状物(6.8mg)。lcms:(m+1)m/z=470.以24%产率,得到(2r,4r)-1-(8-氯-6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物192),作为浅橙色油状物(7.1mg)。lcms:(m+1)m/z=470.实施例193-194反应试剂和条件:i)int-193-48(1.0eq.),胺(2.0eq.),kf(2.5eq.),dmso,125℃,14h,18-21%.(2r,4r)-1-(6-环丙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物193)的合成将dmso(200μl)中的int-193-48(10mg,0.031mmol)、int-193-4(7.4mg,0.037mmol)和kf(4.5mg,0.078mmol)的混合物在125℃加热过夜。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc,利用ch2cl2/meoh(95∶5)纯化。以21%产率得到化合物193,作为作为浅橙色油状物(3.2mg)。lcms:(m+1)m/z=480.以18%产率,得到(2r,4r)-1-(6-环丙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物194),作为浅黄色油状物(3.9mg)。lcms:(m+1)m/z=466.实施例195-197反应试剂和条件i)int-195-46(1.0eq.),胺(1.5eq.),kf(2.5eq.),dmf,125℃,18h,10-22%.(2r,4r)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物195)的合成将dmso(200μl)中的int-195-46(15mg,0.049mmol)、int-195-13(15.6mg,0.074mmol)和kf(7mg,0.123mmol)的混合物在125℃加热过夜。将混合物冷却至室温,用etoac(100ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc,利用ch2cl2/meoh(95∶5)纯化。以22%产率得到化合物195,作为作为浅棕色油状物(4.9mg)。lcms:(m+1)m/z=454.以12%产率,得到(2r,4r)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物196),作为浅棕色油状物(2.6mg)。lcms:(m+1)m/z=454.以10%产率,得到(2r,4r)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物197),作为浅黄色油状物(3.2mg)。lcms:(m+1)m/z=468.实施例198-203反应试剂和条件:i)int-198-49(1.0eq.),胺(1.5eq.),kf(2.0eq.),dipea(3.0eq.),dmso,125℃,18h,50-56%;ii)int-198-50(1.0eq.),nab(och3)4(4.0eq.),pd2(dba)3(2.5%eq.),tbuxphos(5.5%eq.),1,4125℃,2h,6-10%.int-198-50的合成将dmso(1ml)中的int-198-49(100mg,0.28mmol)、实施例195-197中的胺int-195-13(77mg,0.42mmol)、kf(33mg,0.56mmol)和dipea(150μl,0.84mmol)的混合物在125℃加热过夜。冷却至室温后,将混合物用ch2cl2稀释,并用盐水洗涤。将有机相分离,经na2so4干燥,并真空浓缩。将粗品经柱色谱纯化(0~50%etoac的己烷溶液,然后0~10%meoh的ch2cl2溶液)。以54%产率得到产物,作为绿色油状物(77mg)。lcms:(m+1)m/z=504.506.与实施例187-189中的int-187-16反应;79mg,绿色油状物,产率56%。lcms:(m+1)m/z=504,506.与实施例193-194中的int-187-4反应;73mg,绿色固体物,产率50%。lcms:(m+1)m/z=518,520.int-198-51合成前的nab(ome)4的制备将nabh4(0.5g)的meoh(25ml)溶液回流30min。冷却至室温后,将混合物真空浓缩至干,并且没有进一步纯化而使用。int-198-51(a:化合物198,b:化合物199,c:化合物200)和52(a:化合物201,b:化合物202,c:化合物203)的合成将1,4-二氧六环(1.5ml)中的int-198-50(77mg,0.15mmol)、nab(ome)4(96mg,0.61mmol)、pd2(dba)3(3.5g,2.5mol%)和tbuxphos(3.6g,5.5mol%)的混合物在125℃搅拌2h。冷却至室温后,将混合物在ch2cl2和盐水之间进行分配。将有机层经na2so4干燥,并真空浓缩。经prep-tlc,利用etoac∶iproh(97∶3)纯化。以6%产率得到(2r,4r)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物198),作为黄色固体物(4.2mg)。lcms:(m+1)m/z=456;以3%产率得到副产物,(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物201),作为黄色油状物(2.4mg)。lcms:(m+1)m/z=426.以10%产率得到(2r,4r)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物199),作为黄色固体物(7.1mg)。lcms:(m+1)m/z=456.以6%产率得到(2r,4r)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物200),作为黄色固体物(4.2mg)。lcms:(m+1)m/z=470.以5%产率得到(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物202),作为黄色油状物(3.2mg)。lcms:(m+1)m/z=426.以0.8%产率得到(2r,4r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物203),作为黄色油状物(0.5mg)。lcms:(m+1)m/z=440.实施例204反应试剂和条件:i)int-204-49(1.0eq.),int-172-18(1.0eq.),kf(2.0eq),dipea(2.0eq.),dmso,125℃,18h,38%;ii)int-204-53(1.0eq.),nab(och3)4(4.0eq.).pd2(dba)3(2.5%eq.),tbuxphos(5.5%eq.),1,4-二氧六环125℃,2h,8%.(2r,4s)-1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-204-53的合成将dmso(1ml)中的int-204-49(45.2mg,0.127mmol)、实施例172中的胺int-172-18(23.4mg,0.127mmol)、kf(15mg,0.254mmol)和dipea(44μl,0.254mmol)的混合物在125℃加热过夜。冷却至室温后,将混合物用ch2cl2稀释,并用盐水洗涤。将有机相分离,经na2so4干燥,并真空浓缩。将粗品经柱色谱纯化,利用己烷/etoac(0~50%etoac的己烷溶液),然后利用ch2cl2/meoh(0~10%meoh的ch2cl2溶液)作为流动相。以38%产率得到产物int-204-53,作为绿色油状物(24mg)。lcms:(m+1)m/z=505.(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物204)的合成将1,4-二氧六环(1ml)中的int-204-53(24mg,0.047mmol)、nab(ome)4(30mg,0.19mmol)、pd2(dba)3(1g,2.5mol%)和tbuxphos(1mg,5.5mol%)的混合物在125℃搅拌2h。冷却至室温后,将混合物在ch2cl2和盐水之间进行分配。将有机层经na2so4干燥,并真空浓缩。将残留物经prep-tlc纯化,利用etoac∶iproh(97∶3)。以8%产率得到化合物204,作为黄色固体物(1.7mg)。lcms:(m+1)m/z=456.实施例205反应试剂和条件:i)int-205-49(1.0eq.),int-172-20(1.0eq.),kf(2.0eq.),dipea(2.0eq.),dmso,125℃,18h,51%;ii)int-205-54(1.0eq.),nab(och3)4(4.0eq.),pd2(dba)3(2.5%eq.),tbuxphos(5.5%eq.),1,4-二氧六环,125℃,2h,4%.(2s,4s)-1-(6-溴-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺int-205-54的合成将dmso(1ml)中的int-205-49(33.2mg,0.093mmol)、实施例172中的胺int-172-20(17.2mg,0.093mmol)、kf(11mg,0.186mmol)和dipea(32μl,0.186mmol)的混合物在125℃加热过夜。冷却至室温后,将混合物用ch2cl2稀释,并用盐水洗涤。将有机相分离,经na2so4干燥,并真空浓缩。将粗品经柱色谱,利用(0~50%etoac的己烷溶液,然后0~10%meoh的ch2cl2溶液)纯化。以51%产率得到产物int-205-54,作为绿色油状物(24mg)。lcms:(m+1)m/z=504,506.(2s,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物205)的合成将1,4-二氧六环(1ml)中的int-205-54(24mg,0.047mmol)、nab(ome)4(30mg,0.19mmol)、pd2(dba)3(1g,2.5mol%)和tbuxphos(1mg,5.5mol%)的混合物在125℃搅拌2h。冷却至室温后,将混合物在ch2cl2和盐水之间进行分配。将有机层经na2so4干燥,并真空浓缩。将残留物经prep-tlc,利用etoac∶iproh(97∶3)纯化。以4%产率得到化合物205,作为黄色固体物(0.9mg)。lcms:(m+1)m/z=456.实施例206-208反应试剂和条件:i)int-206-50(1.0eq.),nab(ocd3)4(4.0eq.),pd2(dba)3(2.5%eq.),tbuxphos(5.5%eq.),1,4125℃,18h,1-11%.(2r,4r)-1-(8-氟-6-(甲氧基-d3)-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物206)的合成将nabh4(0.1g)的cd3od(5ml)溶液回流30min。冷却至室温后,将混合物真空浓缩至干,并且不需纯化而使用。将1,4-二氧六环(1.5ml)中的int-206-50(70.5mg,0.14mmol)、nab(ocd3)4(95mg,0.56mmol)、pd2(dba)3(3.2g,2.5mol%)和tbuxphos(3.2mg,5.0mol%)的混合物在125℃搅拌过夜。冷却至室温后,将混合物在ch2cl2和盐水之间进行分配。将有机层经na2so4干燥,并真空浓缩。将残留物经prep-tlc,利用etoac∶iproh(97∶3)纯化。以4%产率得到化合物206,作为黄色油状物(2.7mg)。lcms:(m+1)m/z=459.以1%产率,得到(2r,4r)-1-(8-氟-6-(甲氧基-d3)-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物207),作为黄色油状物(0.6mg)。lcms:(m+1)m/z=459.以11%产率,得到(2r,4r)-1-(8-氟-6-(甲氧基-d3)-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物208),作为黄色油状物(6mg)。lcms:(m+1)m/z=473.实施例209-211反应试剂和条件:i)int-209-50(1.0eq.),iprmgcl(3.0eq.),pd(oac)2(0.1eq.),p(tbu)3hbf4(0.12eq.),znbr2(0.5eq.),thf,25℃,2h,8-13%.(2r,4r)-1-(8-氟-6-异丙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物209)的合成向int-209-50(70.5mg,0.14mmol)、pd(oac)2(3.1mg,0.014mmol)、p(tbu)3hbf4(4.9mg,0.017mmol)、znbr2(70μl,1mthf溶液)在thf(1.5ml)的混悬液中,加入2miprmgcl溶液(0.21ml,0.42mmol),并将混合物在室温搅拌2h。然后,将混合物倒入冰-水中,并将产物用etoac(3x)萃取。将合并的有机层用1%aq.hcl和盐水洗涤,na2so4干燥,并真空浓缩。将残留物经prep-tlc,利用etoac∶iproh(97∶3)和ch2cl2∶meoh(96∶4)纯化。以8%产率,得到化合物209,作为黄色固体物(5.4mg)。lcms:(m+1)m/z=468.以8%产率,得到(2r,4r)-1-(8-氟-6-异丙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物210),作为黄色固体物(5.2mg)。lcms:(m+1)m/z=468.以13%产率,得到(2r,4r)-1-(8-氟-6-异丙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物211),作为黄色油状物(7.4mg)。lcms:(m+1)m/z=482.实施例212-213反应试剂和条件i)int-212-55(1.0eq.)int-172-16(1.5eq.)kf(2.0eq.)dipea(3.0eq.),dmso,125℃,18h,48%;ii)int-212-56(1.0eq.),zn(cn)2(1.0eq.),zn(0.1eq.),pd(pph3)4(0.1eq),thf/dmf(1∶1,v/v),80℃,18h,2%(对于化合物212)and58%(forint-212-57);iii)int-21-57(1.0eq.),etb(oh)2(1.4eq.),pcy3(0.1eq.),k3po4(3.6eq.),pd(oac)2(5%eq.)甲苯h2o,100℃,18h,18%.(2r,4r)-1-(6-溴-8-碘-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺int-212-56的合成将dmso(2ml)中的int-212-55(150mg,0.32mmol)、实施例172中的胺int-172-16(89mg,0.48mmol)、kf(37mg,0.64mmol)和dipea(0.17ml,0.96mmol)的混合物在125℃加热过夜。冷却至室温后,将混合物用ch2cl2稀释,并用盐水洗涤。将有机相分离,经na2so4干燥,并真空浓缩。将粗品经柱色谱,利用己烷/etoac(0~50%etoac的己烷溶液),然后利用ch2cl2/meoh(0~10%meoh的ch2cl2溶液)纯化。以48%产率得到产物,作为深绿色油状物(94.3mg)。lcms:(m+1)m/z=612,614.4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-(((r)-四氢呋喃-3-基)氨基)哌啶-1-基)喹啉-6,8-二腈(化合物212)和57的合成将int-212-56(94.3mg,0.154mmol)、zn(cn)2(18.0mg,0.154mmol)、zn粉(1.0mg,0.0154mmol)和pd(pph3)4(17.8mg,0.0154mmol)在thf/dmf(2ml,1∶1,v/v)中的混合物于80℃搅拌过夜。冷却至室温后,将混合物经硅藻土过滤,并真空浓缩滤液。将残留物经prep-tlc,利用etoac∶iproh(98∶2)纯化,以生产化合物212,作为黄色固体物(1.4mg,产率2%),和以58%产率生产57,作为黄色固体物(45.6mg)。lcms:(m+1)m/z=511,513。6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-(((r)-四氢呋喃-3-基)氨基)哌啶-1-基)喹啉-8-腈的合成(化合物213)将甲苯(2ml)和h2o(40μl)中的int-212-57(45.6mg,0.089mmol)、etb(oh)2(9.2g,0.125mmol)、p(cy)3(2.5mg,0.0089mmol)、pd(oac)2(1.0mg,0.0045mmol)和k3po4(68mg,0.32mmol)的混合物在100℃搅拌和加热过夜。冷却至室温后,将混合物经硅藻土过滤。将滤液真空浓缩,并经prep-tlc,利用etoac∶iproh(98∶2),然后利用ch2cl2/meoh(97∶3)纯化。以18%产率得到产物化合物213,作为黄色固体物(7.4mg)。lcms:(m+1)m/z=461.实施例214-215反应试剂和条件:i)int-214-58(1.0eq.),et3b(1.5eq.),k2co3(2.0eq.),pd(pph3)4(5%eq.),thf/dmf(1∶1,v/v),80℃,1.5h,66%;ii)int-214-59(1.0eq.),nis(1.05eq.),acoh,rt,1.5h,88%;iii)int-214-60(1.0eq.),pocl3,100℃,1h,44%;iv)int-214-61(1.0eq),胺:(1.5eq.),kf(2.0eq.)dipea(3.0eq.),dmso,125℃,18h,32-41%;v)int-214-62(1.0eq)zn(cn)2(1.5eq.),zn(0.1eq.),pd(pph3)4(0.1eq),dmf,80℃,18h,13-17%.int-214-59的合成在室温,向thf/dmf(60ml,1∶1v/v)中的int-214-58(6.0g,28.0mmol)、k2co3(7.7g,56.0mmol)和pd(pph3)4(1.6g,1.4mmol)的混合物中,加入1met3b的thf溶液(42ml,42.0mmol)。然后,将混合物在80℃加热1.5h。冷却至室温后,将混合物经硅藻土过滤。将滤液真空浓缩,并经柱色谱纯化,利用己烷/etoac(0~20%etoac的己烷溶液)作为流动相。以66%产率得到产物int-214-59,作为黄色固体物(3.0g)。lcms:(m+1)m/z=164.int-214-60的合成向int-214-59(2.5g,15.6mmol)的乙酸(50ml)混合物中,一次性加入nis(3.7g,16.4mmol),并将混合物在室温搅拌1.5h。将混合物用etoac(500ml)稀释,并用盐水(3x)和饱和nahco3水溶液(2x)洗涤。将有机相用na2so4干燥,并真空浓缩。将产物经柱色谱纯化,利用己烷/etoac(0~30%etoac的己烷溶液)作为流动相。以88%产率得到产物,作为红色油状物(4.0g)。lcms:(m+1)m/z=290.int-214-61的合成将pocl3(14ml)中的int-214-60(2.09g,7.23mmol)和int-214-25(1.13g,7.95mmol)的混合物在100℃搅拌1h。冷却至室温后,真空除去过量的pocl3。在0℃,向残留物中加入h2o,并将混合物在0℃搅拌10min。将沉淀的粗氯喹啉int-214-61过滤,用h2o洗涤,并减压干燥。将固体物经柱色谱纯化,利用0~20%etoac的己烷溶液作为流动相。以44%产率得到产物,作为黄色固体物(1.33g)。int-214-62的合成将dmso(2ml)中的int-214-61(75mg,0.18mmol)、int-214-13(50mg,0.27mmol)、kf(21mg,0.36mmol)和dipea(94μl,0.54mmol)的混合物在125℃加热过夜。冷却至室温后,将混合物用ch2cl2稀释,并用盐水洗涤。将有机相经na2so4干燥,并真空浓缩。将粗品经柱色谱纯化,利用己烷/etoac(0~50%etoac的己烷溶液),然后ch2cl2/meoh(0~10%meoh的ch2cl2溶液)作为流动相。以41%产率得到产物int-214-62a,作为深棕色油状物(41.4mg)。lcms:(m+1)m/z=562.与胺int-214-4反应,得到int-214-62b(33.1mg,产率32%),作为深棕色油状物。lcms:(m+1)m/z=576.6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-(((s)-四氢呋喃-3-基)氨基)哌啶-1-基)喹啉-8-腈(化合物214)的合成将dmf(2ml)中的int-214-62(41.4mg,0.074mmol)、zn(cn)2(13.0mg,0.111mmol)、zn粉(0.5mg,0.0074mmol)和pd(pph3)4(8.6mg,0.0074mmol)的混合物于80℃搅拌过夜。冷却至室温后,将混合物经硅藻土过滤,并真空浓缩滤液。将残留物经prep-tlc,利用etoac∶iproh(98∶2),然后利用ch2cl2/meoh(97∶3)纯化。以17%产率得到化合物214,作为黄色固体物(6.0mg)。lcms:(m+1)m/z=461.以29%产率得到6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-((四氢-2h-吡喃-4-基)氨基)哌啶-1-基)喹啉-8-腈(化合物215),作为黄色固体物(7.9mg)。lcms:(m+1)m/z=475.实施例216反应试剂和条件:i)int-216-66(1.0eq.),胺(1.2eq.),tmp/dmso(1∶1,v/v),150℃,18h,8%.(2r,4s)-1-(6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物216)的合成将2,2,6,6-四甲基哌啶/dmso(1.5ml,1∶1v/v)中的int-216-66(30mg,0.104mmol)和7(23mg,0.125mmol)的混合物在150℃加热过夜。冷却至室温后,将混合物在ch2cl2和盐水之间进行分配。有机层经na2so4干燥,并真空浓缩。将残留物经制备型-tlc(ch2cl2∶meoh=96∶4)纯化。以8%产率得到产物化合物216,作为棕色油状物(3.7mg)。lcms:(m+1)m/z=450.实施例217反应试剂和条件:i)int-217-66(1.0eq.),胺(1.2eq.),tmp/dmso(1∶1,v/v),150℃,18h,6%.(2r,4r)-1-(6-乙基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物217)的合成将2,2,6,6-四甲基哌啶/dmso(1.5ml,1∶1v/v)中的int-217-66(30mg,0.104mmol)和int-217-4(23mg,0.125mmol)的混合物在150℃加热过夜。冷却至室温后,将混合物在ch2cl2和盐水之间进行分配。有机层经na2so4干燥,并真空浓缩。将残留物经制备型-tlc(ch2cl2/meoh(96∶4))纯化。以6%产率得到产物化合物217,作为棕色油状物(2.7mg)。lcms:(m+1)m/z=450.实施例218-220反应试剂和条件:i)int-218-67(1.0eq.),胺(1.2eq.),kf(2.5eq.),dipea(2.0eq.),dmf,125℃,18h,13%.(2r,4r)-1-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物218)的合成将dmso(200μl)中的int-218-67(15mg,0.051mmol)和int-172-4(12.3mg,0.062mmol)、kf(7mg,0.128mmol)和dipea(18μl,0.102mmol)的混合物在125℃加热过夜。将混合物冷却至室温,用etoac(60ml)稀释,并用1mnaoh溶液(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经prep-tlc,利用ch2cl2/meoh(95∶5)纯化。以13%产率得到化合物218,作为作为浅黄色油状物(3.1mg)。lcms:(m+1)m/z=454.以15%产率得到(2r)-1-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺(化合物219),作为浅黄色油状物(3.3mg)。lcms:(m+1)m/z=440.以13%产率得到(2r)-1-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺(化合物220),作为浅黄色油状物(3.0mg)。lcms:(m+1)m/z=440.根据对于化合物219-220所述的方法,用一种适当的立体化学和官能团代替胺int-218-4,得到化合物219a和220a。实施例221-223反应试剂和条件:i)int-221-29(1.0eq.),1,4-二氧杂-8-氮杂螺[4.5]癸烷(2.0eq.),dipea(2.0eq.),iproh,120℃,过夜,23%;ii)int-221-69(1.0eq.),h2so4(10%aq.),thf,40℃,2h,62%;iii)int-221-70(1.0eq.),胺(2.0eq.),nabh(oac)3(2.0eq.),acoh(2.0eq.),dipea(2.0eq.),1,2-二氯乙烷,rt,过夜,76-95%.int-221-69的合成将iproh(4ml)中的int-221-29(210mg,0.669mmol)、1,4-二氧杂-8-氮杂螺[4,5]癸烷(190mg,1.33mmol)和dipea(233μl,1.33mmol)的混悬液在120℃加热过夜。冷却至室温后,将混合物减压浓缩并经柱色谱纯化(己烷/etoac),以23%产率得到缩酮int-221-69,作为黄色固体物(65mg)。lcms:(m+1)m/z=419.int-221-70的合成将缩酮int-221-69(60mg,0.143mmol)的thf/10%aq.h2so4(1∶1,1.5ml)溶液在40℃搅拌2h。冷却至室温后,将混合物用饱和naoh水溶液中和,并用etoac(2x)萃取。将有机层经na2so4干燥,并减压浓缩,以62%产率得到酮int-221-70,作为黄色固体物(33mg),没有进一步纯化,将其用于下一步骤。lcms:(m+1)m/z=367.化合物221-223的合成将1,2-二氯乙烷中的酮int-221-70(8mg,0.021mmol)、4-氨基四氢吡喃(4.3mg,0.043mmol)、dipea(7.4μl,0.043mmol)、nabh(oac)3(9mg,0.043mmol)和acoh(2.4μl,0.043mmol)的混合物在室温搅拌过夜。将混合物经硅藻土过滤,减压浓缩,并经prep-tlc,利用ch2cl2/meoh(95∶5)纯化,以91%产率得到1-(6-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物221),作为黄色固体物(8.9mg)。lcms:(m+1)m/z=460.以95%产率得到(s)-1-(6-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物222),作为浅黄色固体物(9.0mg)。lcms:(m+1)m/z=446.以76%产率得到(s)-1-(6-氯-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-3-基)哌啶-4-胺(化合物223),作为浅黄色油状物(7.4mg)。lcms:(m+1)m/z=460.实施例224-225反应试剂和条件:i)int-224-67(1.0eq.),1,4-二氧杂-8-氮杂螺[4.5]癸烷(2.0eq.),dipea(2.0eq.),etoh,120℃,过夜,84%;ii)int-224-71(1.0eq.),h2so4(10%aq.),thf,50℃,2h,87%;iii)int-224-72(1.0eq.),胺(1.2eq.),nabh(oac)3(2.0eq.),acoh(2.0eq.),dipea(2.0eq.),1,2-二氯乙烷,rt,过夜50-62%.int-224-71的合成在室温,向氯喹啉int-224-67(210mg,0.75mmol)和dipea(0.26ml,1.50mmol)的etoh(5ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(0.20ml,1.50mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩并经柱色谱纯化,(0~20%etoac的己烷溶液)作为流动相。以84%产率得到产物int-224-71,作为浅黄色固体物(251mg)。lcms:(m+1)m/z=399.int-224-72的合成在室温,向int-224-71(246mg,0.62mmol)的thf(2.5ml)溶液中,加入10%aq.h2so4(5ml)。然后,将混合物在50℃搅拌1h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(2×20ml)。将合并的有机层经na2so4干燥,并真空浓缩。将产物经柱色谱纯化,(0~66%etoac的己烷溶液)作为流动相,以87%产率得到产物int-224-72,作为黄色固体物(189mg)。lcms:(m+1)m/z=355.(r)-1-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物224)的合成将1,2-二氯乙烷(1ml)中的int-224-72(10.0mg,0.028mmol)、(r)-3-氨基四氢呋喃盐酸盐(4.2mg,0.034mmol)和dipea(10μl,0.056mmol)的混合物在室温搅拌10min。向混合物中,加入nabh(oac)3(12.0mg,0.056mmol)和acoh(3μl,0.056mmol)。将得到的混合物在室温下搅拌过夜。然后,将混合物经硅藻土过滤,并将滤液真空浓缩。将残留物经prep-tlc,利用ch2cl2/meoh(96∶4)纯化。以62%产率得到产物化合物224,作为黄色固体物(7.4mg)。lcms:(m+1)m/z=426.以50%产率得到(s)-1-(8-氟-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物225),作为黄色固体物(5.9mg)。lcms:(m+1)m/z=426.实施例226-227反应试剂和条件:i)int-226-33(1.0eq.),1,4-二氧杂-8-氮杂螺[4.5]癸烷(2.0eq.),dipea(2.0eq.),etoh,120℃,过夜,77%;ii)int-226-73(1.0eq.),h2so4(10%aq.),thf,50℃,2h,59%;iii)int-226-74(1.0eq.),胺(1.2eq.),nabh(oac)3(2.0eq.),acoh(2.0eq.),dipea(2.0eq.),1,2-二氯乙烷,rt,过夜67-79%.int-226-73的合成在室温,向氯喹啉int-226-33(170mg,0.47mmol)和dipea(0.16ml,0.94mmol)的etoh(5ml)混悬液中,加入1,4-二氧杂-8-氮杂螺[4,5]癸烷(0.12ml,0.94mmol)。然后,将混合物在120℃加热过夜。冷却至室温后,将混合物浓缩并经柱色谱纯化,(0~20%etoac的己烷溶液)作为流动相。以77%产率得到产物int-226-73,作为红色油状物(170mg)。lcms:(m+1)m/z=469.int-226-74的合成在室温,向int-226-73(170mg,0.36mmol)的thf(2ml)溶液中,加入10%aq.h2so4(3ml)。然后,将混合物在50℃搅拌2h。冷却至室温后,将混合物用饱和na2co3水溶液中和,并用etoac萃取(2×20ml)。将合并的有机层经na2so4干燥,并真空浓缩。将产物经柱色谱纯化,(0~66%etoac的己烷溶液)作为流动相。以59%产率得到产物int-226-74,作为红色油状物(91mg)。lcms:(m+1)m/z=425.1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-6-(三氟甲氧基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物226)的合成将1,2-二氯乙烷(1ml)中的int-226-74(15.0mg,0.035mmol)、4-氨基四氢吡喃(4.2mg,0.042mmol)和dipea(12μl,0.070mmol)的混合物在室温搅拌10min。向混合物中,加入nabh(oac)3(14.8mg,0.070mmol)和acoh(4μl,0.070mmol)。将得到的混合物在室温搅拌过夜。然后,将混合物经硅藻土过滤,并将滤液真空浓缩。将残留物经prep-tlc,利用ch2cl2/meoh(96:4)纯化。以67%产率得到产物化合物226,作为棕色固体物(11.9mg)。lcms:(m+1)m/z=510.以79%产率得到(r)-1-(8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)-6-(三氟甲氧基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物227)(13.9mg)。lcms:(m+1)m/z=496.实施例228(r)-1-(8-氟-6-(甲氧基-d3)-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物228)的合成步骤1:按照int-33-14j的制备,其中用kb(ocd3)4代替kb(och3)4,经硅胶色谱纯化(0-60%etoac/己烷)后,得到int-228-1,作为白色固体物。lcms:m/z418(m+h).步骤2-3:根据上文对于化合物34公开的方法,并利用相应的氘-取代的中间体,得到化合物228,作为白色固体物。lcms:m/z445(m+h);保留时间:3.55min(方法2)。实施例229根据化合物197合成的通用方法,利用适当的手性胺,得到化合物229,作为白色固体物;lcms:m/z468(m+h).实施例230-235利用实施例195-197或236-237中公开的方法,利用胺和适当立体化学的烷基化试剂,得到化合物230-235。实施例236-237236a和236b:(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺和(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺的混合物和237a和237b:(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺和(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺的混合物反应试剂和条件:i)kb(ome)4(3equiv.),pd2(dba)3(0.05equiv),tbuxphos(0.1equiv.),二氧六环100℃,2h;ii)kf(2.5equiv.),dmso,130℃;iii)tfa,dcm;iv)dipea(4equiv.),ch3cn,uw,120℃;iv)dipea(4equiv.),ch3cn,uw,120℃.根据int-33-14j的通用方法,由int-1-1c合成int-236-1。1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-反式-2-甲基哌啶-4-基)氨基甲酸叔丁酯(int-236-3)的合成向微波瓶中,填充5-(2-氯-8-氟-6-甲氧基-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑(int-236-1)(0.143mg,0.45mmol)、外消旋n-[(反式)-2-甲基哌啶-4-基]氨基甲酸叔丁酯(int-236-2,0.199g,0.929mmol)、氟化钾(67.5mg,1.16mmol)、无水dmso(1.2ml)和二异丙基乙胺(0.21ml,1.16mmol)。将混合物置于氮气气氛下,并在130℃下加热15小时。冷却至室温后,将混合物在乙酸乙酯与饱和碳酸氢钠水溶液中进行分配。将有机萃取物依次用碳酸氢钠和盐水溶液洗涤,经硫酸钠干燥,并浓缩至300mg。将粗产物经柱色谱纯化,用0-40%ea/己烷梯度从12g硅胶洗脱,得到目标产物,作为黄色油状物(140mg,61%产率)。lcms:(m+1)m/z=486.1.1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-反式-2-甲基哌啶-4-胺(int-236-4)的合成向0℃冷却的1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-反式-2-甲基哌啶-4-基)氨基甲酸叔丁酯(int-236-3,135mg,0.278mmol)的5mldcm溶液中,加入2.5mltfa。将混合物在室温搅拌1.5hr,浓缩为橙色泡状物,并在等体积的半饱和na2co3溶液和etoac之间进行分配。将有机溶液用na2co3溶液洗涤两次,经硫酸钠干燥,并浓缩,得到int-236-4,作为黄色固体物(99mg,产率94%)。lcms:(m+1)m/z=386.2.化合物236a(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺和236b(2s,4r)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((r)-四氢呋喃-3-基)哌啶-4-胺的合成向微波管中,填充1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-反式-2-甲基哌啶-4-胺(int-236-4,55mg,0.14mmol)、4-甲基苯-1-磺酸(3s)-四氢呋喃-3-基酯(int-172-14,140mg,0.56mmol)、二异丙基乙胺(97μl,0.56mmol)和ch3cn(0.9ml)。将混合物在微波反应器中于120℃下加热12小时。将混合物在etoac与na2co3水溶液中进行分配,用硫酸钠干燥有机部分,并浓缩至225mg橙色油状物。将粗产物经柱色谱纯化,用0-60%etoac/己烷梯度,随后用0-10%meoh/dcm梯度从硅胶洗脱,得到非对映异构体的混合物,作为黄色固体物(40mg,63%产率)。可利用phenomenexlux5μcellulose-2柱子(250mmx4.6mm,己烷:乙醇:dea(80:20:1),1ml/min,保留时间:15.7min,17.5min,将混合物分离,得到单独的非对映体。lcms:(m+1)m/z=356.1.化合物237a(2r,4s)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺和化合物237b(2s,4r)-1-(8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-2-甲基-n-((s)-四氢呋喃-3-基)哌啶-4-胺的合成向微波管中,填充1-[8-氟-6-甲氧基-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基]-反式-2-甲基哌啶-4-胺(int-236-4,58mg,0.15mmol)、4-甲基苯-1-磺酸(3r)-四氢呋喃-3-基酯(int-172-11,146mg,0.56mmol)、dipea(0.105ml,0.56mmol)和ch3cn(0.9ml)。将混合物在微波反应器中于120℃下加热12小时。将混合物在etoac与na2co3水溶液之间进行分配,用硫酸钠干燥有机部分,并浓缩至170mg橙色油状物。将粗产物经柱色谱纯化,用0-60%etoac/己烷梯度,随后用0-10%meoh/dcm梯度洗脱,得到非对映异构体的混合物,作为黄色固体物(38mg,产率56%)。可利用phenomenexlux5μcellulose-4柱子(250mmx4.6mm,己烷:乙醇:dea(73:27:0.1),1ml/min,保留时间:7.2min,8.9min,将混合物分离,得到单独的非对映体。lcms:(m+1)m/z=356.1.实施例238-239根据上文公开的化合物228的方法,并在步骤3中应用适当的胺,制备化合物238和239。化合物238:黄色固体物;lcms:m/z459(m+h);保留时间:3.71min(方法2)。化合物239:白色固体物;lcms:m/z429(m+h);保留时间:3.90min(方法2)。实施例240步骤1:向5-(6-溴-2-氯-8-氟-4-甲基喹啉-3-基)-3-甲基-1,2,4-二唑(357mg,1.00mmol)和双(三苯基膦)氯化钯(ii)(68mg,88umol)在thf(2ml)中的混悬物中,加入(甲基-d3)碘化锌(ii)混悬液(7.0ml,ref:doi:10.1039/c7cc06106d),并将反应物在60℃搅拌4h。用柠檬酸溶液(5%aq.)淬灭反应,并用乙酸乙酯萃取两次。将有机层合并,用盐水洗涤,经na2so4干燥,并减压浓缩。将残留物经硅胶柱色谱(etoac/hex)纯化,得到目标产物,作为白色固体物(153mg,产率52%)。lcms(esi):m/z295(m+h).步骤2:类似于化合物174,但是用5-(2-氯-8-氟-4-甲基-6-(甲基-d3)喹啉-3-基)-3-甲基-1,2,4-二唑代替5-(2,8-二氯-4,6-二甲基喹啉-3-基)-3-甲基-1,2,4-二唑,得到化合物240,作为白色固体物。lcms:m/z457(m+h).实施例241-242根据化合物240的方法,得到化合物241,作为白色固体物;lcms:m/z443(m+h);保留时间:3.62min(方法2)。得到化合物242,作为白色固体物;lcms:m/z443(m+h);保留时间:3.74min(方法2)。实施例243根据化合物197的通用合成方法,利用适当的手性胺,得到化合物243,作为白色固体物;lcms:m/z468(m+h).实施例244利用实施例259-260中公开的方法,用适当的手性胺int-259-13和int-36-14h,得到化合物244。实施例245-2484,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-(((四氢-2h-吡喃-4-基)甲基)氨基)哌啶-1-基)喹啉-8-腈(化合物245)的合成.将dmso(2ml)中的int-245-1(200mg,0.67mmol)、int-245-2(172mg,0.804mmol)和kf(97mg,1.67mmol)的混合物在125℃加热过夜。将混合物冷却至室温后,用etoac(150ml)稀释,并用盐水(3x)洗涤。将有机相经na2so4干燥,减压浓缩,并将产物经柱色谱纯化,利用己烷/etoac(7:3)。以56%产率得到产物,作为黄色固体物(180mg)。lcms:(m+1)m/z=477.将ch2cl2(5ml)中的int-245-3(170mg,0.356mmol)和tfa(550μl,7.13mmol)的混合物在室温搅拌1h。将混合物减压浓缩。将粗品再溶解于etoac中,并用2mnaoh(2x)洗涤。将水相用etoac(2x)萃取。将合并的有机相经na2so4干燥,减压浓缩,并且没有进一步纯化而使用产物。以95%产率得到int-245-4,作为黄色固体物。lcms:(m+1)m/z=377.将dce(200μl)中的胺int-245-4(10mg,0.026mmol)和int-245-5(3mg,0.026mmol)的混合物在室温搅拌1h,随后加入nabh(oac)3(11mg,0.052mmol)和acoh(2.9μl,0.052mmol)。将得到的混合物在室温搅拌过夜。将混合物用etoac(50ml)稀释,并用2mnaoh(1x)洗涤。将有机层经na2so4干燥,并浓缩。将残留物经prep-tlc纯化,利用ch2cl2/meoh(95:5)。以49%产率得到化合物245,作为黄色固体物(6.1mg)。lcms:(m+1)m/z=475.4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-((((r)-四氢呋喃-2-基)甲基)氨基)哌啶-1-基)喹啉-8-腈(化合物246)的合成将乙腈(150μl)中的int-245-4(10mg,0.026mmol)、int-245-6(13.3mg,0.052mmol)和dipea(9.1μl,0.052mmol)的混合物在80℃加热过夜。将混合物减压浓缩,并经prep-tlc,利用ch2cl2/meoh(95:5)纯化。以53%产率得到化合物246,作为黄色油状物(6.3mg)。lcms:(m+1)m/z=461.4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)-2-((2r,4r)-2-甲基-4-((((s)-四氢呋喃-2-基)甲基)氨基)哌啶-1-基)喹啉-8-腈(化合物247)的合成用相同方式合成化合物247,并以50%产率得到,作为黄色油状物(6.0mg)。lcms:(m+1)m/z=461.2-((3s,4r)-3-氟-4-((四氢-2h-吡喃-4-基)氨基)哌啶-1-基)-4,6-二甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-8-腈(化合物248)的合成将etoh(250μl)中的int-245-1(20mg,0.067mmol)、int-245-8(34.4mg,0.08mmol)和dipea(47μl,0.268mmol)的混合物在125℃加热过夜。将混合物减压浓缩,并经prep-tlc,利用ch2cl2/meoh(95:5)纯化。以46%产率得到化合物248,作为黄色固体物(14.3mg)。lcms:(m+1)m/z=465.实施例249(r)-3-(5-甲基-1,2,4-二唑-3-基)-2-(4-((四氢-2h-吡喃-3-基)氨基)哌啶-1-基)-6-(三氟甲氧基)喹啉-8-腈(化合物249)的合成将1,2-二氯乙烷中的酮int-85-43(5mg,0.012mmol)、3-(r)-氨基四氢呋喃hcl(3.3mg,0.024mmol)、dipea(4.2μl,0.024mmol)、nabh(oac)3(5mg,0.024mmol)和acoh(1.3μl,0.024mmol)的混合物在室温搅拌过夜。将混合物经硅藻土过滤,减压浓缩,并经prep-tlc(ch2cl2:meoh=95:5)纯化,得到化合物249(4.1mg,产率69%),作为黄色固体物。lcms:(m+1)m/z=503.实施例2501-(6-乙基-8-氟-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-4-基)哌啶-4-胺(化合物250)的合成将1,2-二氯乙烷(0.4ml)中int-141-8(8mg,0.022mmol)、4-氨基四氢吡喃(3.4mg,0.034mmol)、nabh(oac)3(7.2mg,0.034mmol)和acoh(1.9μl,0.13mmol)的混合物在rt搅拌3h。将混合物直接经制备-tlc(ch2cl2:meoh=95:5)纯化,得到化合物250,作为黄色固体物(8.1mg,产率82%)。lcms:(m+1)m/z=440.实施例251(r)-1-(6-乙基-8-氟-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物251)的合成将1,2-二氯乙烷(0.4ml)中的int-141-8(10mg,0.028mmol)、(r)-四氢呋喃-3-胺盐酸盐(4.2mg,0.034mmol)和dipea(5.9μl,0.11mmol)的混合物在rt搅拌10min。向混合物中,加入nabh(oac)3(8.9mg,0.042mmol)和acoh(2.3μl,0.042mmol)。将得到的混合物在rt搅拌3h。将反应混合物直接经制备型-tlc(ch2cl2:meoh=95:5)纯化,得到化合物251,作为黄色固体物(8.1mg,产率68%)。lcms:(m+1)m/z=426.实施例252-253用与化合物251的类似方式,合成(s)-1-(6-乙基-8-氟-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢呋喃-3-基)哌啶-4-胺(化合物252),并以82%产率作为黄色固体物(9.8mg)得到。lcms:(m+1)m/z=426.用与化合物251的类似方式,合成(s)-1-(6-乙基-8-氟-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-(四氢-2h-吡喃-3-基)哌啶-4-胺(化合物253),并以84%产率作为黄色固体物(10.4mg)得到。lcms:(m+1)m/z=440.实施例254化合物254的合成根据合成化合物170的方法,用6-溴-3-(3-甲基-1,2,4-二唑-5-基)-2-(1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-基)喹啉-8-腈代替6-溴-4-甲基-3-(5-甲基-1,2,4-二唑-3-基)-2-(1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-基)喹啉-8-腈,得到化合物254(7.6mg,最后一步产率40%)。lcms(esi):m/z435(m+h);保留时间:1.86min(方法1)。实施例255根据化合物85所述的方法,用适当的甲基酮代替起始的醛int-85-38,得到化合物255。实施例256-258根据化合物174-176所述的方法,用适当立体化学的胺代替胺int-174-4,得到化合物256-258。实施例259和260(3s,4r)-3-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-259-13的合成将dce(2ml)中的int-259-10(100mg,0.46mmol)、int-259-11(86μl,0.93mmol)、nabh(oac)3(197mg,0.93mmol)和acoh(51.6μl,0.93mmol)的混合物在室温搅拌过夜。将混合物用1mnaoh淬灭,并用etoac(4x)萃取。将有机层经na2so4干燥,并浓缩。经柱色谱纯化,用ch2cl2/meoh梯度洗脱,得到化合物int-259-12,作为白色固体物(122mg,产率89%)。lcms:(m+1)m/z=299.将ch2cl2(2ml)中的int-259-12(120mg,0.402mmol)和tfa(615μl,3.61mmol)的混合物在室温搅拌1h。将混合物减压浓缩。没有进一步纯化而使用产物int-259-13(白色固体物)。lcms:(m+1)m/z=199.(3r,4s)-3-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺int-259-16的合成用与int-259-13类似的方式,用int-259-14代替int-259-10,合成int-259-16,并作为白色固体物得到(产率87%,两步).lcms:(m+1)m/z=199.(3s,4r)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-3-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺化合物259的合成将iproh(250μl)中的int-142-11(15mg,0.049mmol)、int-259-13(31mg,0.074mmol)和dipea(42μl,0.245mmol)的混合物在125℃加热过夜。将混合物减压浓缩,并用prep-tlc纯化,利用ch2cl2/meoh(95:5)作为洗脱剂。以31%产率得到化合物259,作为黄色固体物(7.1mg)。(m+1)m/z=468.(3r,4s)-1-(6-乙基-8-氟-4-甲基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-3-甲基-n-(四氢-2h-吡喃-4-基)哌啶-4-胺化合物260的合成利用化合物259中所用的相同条件,由int-142-11和int-259-16合成化合物260,并以36%产率得到,作为黄色固体物(8.3mg)。lcms:(m+1)m/z=468.实施例261-265根据化合物33-39和198-203中所述的通用方法,在酯化步骤利用环丙醇代替kb(ome)4,和适当的胺,以生产目标产物,得到化合物261-265。实施例b-1体外活性在下文的实验数据中,利用1-(6-乙基-3-(3-甲基-1,2,4-二唑-5-基)喹啉-2-基)-n-((3-甲基氧杂环丁-3-基)甲基)哌啶-4-胺(化合物a)、pf-04455242和ly2456302作为参考化合物。利用美国专利号9,682,966中所述的合成方法制备化合物a。pf-04455242是wo2009/156889中公开的已知的kor拮抗剂。ly2456302是wo2009/094260中公开的已知的kor拮抗剂。kor拮抗剂测试用于oprk1拮抗剂测试的细胞系稳定表达下述元件:具有7-氨基酸连接体的oprk1受体的羧基末端,随后tev蛋白酶裂解位点和gal4-vp16融合蛋白。细胞系还表达b-抑制蛋白-2-tev蛋白酶融合蛋白,并包含由uas反应元件和b-内酰胺酶(bla)报道基因构成的报道构件。一旦受体活化,g-蛋白受体激酶(grk)将细胞内特定残基磷酸化,并且所述诱导b-抑制蛋白2-tev蛋白酶的募集。tev蛋白酶识别和裂解tev位点,释放gal4-vp16融合蛋白,然后其转移至细胞核内。gal4-v16结合至uas元件,驱动b-内酰胺酶基因表达。用可渗透细胞的荧光底物ccf4-am检测b-内酰胺酶表达。所述底物由通过b-内酰胺环结合至荧光素的香豆素构成。无b-内酰胺酶时,具有405nm光的染料激发产生自香豆素至荧光素的fret,以及绿光(最大525nm)发射。底物的b-内酰胺酶裂解将香豆素荧光团自荧光素分离,并且405nm激发产生蓝色(最大460nm)发射。经蓝/绿发射比率检测所述测试。将oprk1tangou2os细胞培养于生长培养基中(mccoy’s5a培养基,10%透析的fbs,非必需氨基酸,25mmhepes,1mm丙酮酸钠,青霉素/链霉素)。将二百万细胞添加至有30ml生长培养基的t175烧瓶中,并在37℃/5%co2下温孵4天,在所述时间点,它们是~70-90%融合的。经抽吸除去生长培养基,向烧瓶中加入5ml0.25%胰蛋白酶/edta,并轻轻洗涤细胞。然后,经抽吸除去胰蛋白酶。让细胞聚集,并通过轻敲烧瓶而脱离。将细胞悬浮于测试培养基中(高糖dmem,具有1%炭葡聚糖处理的(charcoaldextranstripped)fbs、非必需氨基酸、25mmhepes、1mm丙酮酸钠、青霉素/链霉素),研磨,计数,并通过离心而成丸。将细胞以160万细胞/ml再悬浮,并将10μl加入到黑色透明底的384-孔测试板(greinerpartnumber788092)的各孔中。将测试板置于增湿盒中,并在37℃/5%co2下孵育16-24小时。将溶解于dmso中的化合物依次用dmso在384-孔聚丙烯板中稀释。利用pintools,将50nl各化合物的稀释液加至测试板的各孔中。对照孔接受50nldmso。将板子放回培养器中,保持30分钟。用化合物预培养后,向测试板的化合物孔中加入50nl600nm(-)-u-50,488(激动剂激发)。对照孔中接受50nl5.6um(-)-u-50,488(100%响应对照)、50nl600nm(-)-u-50,488(ec80对照)或50nldmso(0%响应对照)。将测试板放回37℃/5%co2的培养器中,保持4小时。然后,将测试板从培养器中移出,并向各孔中加入2.5ulliveblazerccf4-am底物染料(invitrogen)。然后,将测试板在室温置于台子上,用铝箔覆盖,以避光,保持2小时。然后,将板子在荧光板读数器上,用405nm的激发波长,以及460nm和525nm的发射波长读数。利用蓝/绿的发射比率计算结果。利用下述等式计算抑制百分数,ic50是实现50%抑制需要的化合物浓度:对于外周κ拮抗剂有效且具有最小副作用,期望的是其不与其他阿片样受体以任何实质性程度相互作用。因此,利用δ和μ阿片样受体拮抗剂测试来评价μ或δ受体的活化。oprδ拮抗剂测试用于oprδ拮抗剂测试的细胞系稳定表达下述元件:具有7-氨基酸连接体的oprδ受体的羧基末端,以及tev蛋白酶裂解位点和gal4-vp16融合蛋白。细胞系还表达b-抑制蛋白-2-tev蛋白酶融合蛋白,并包含由uas反应元件和b-内酰胺酶(bla)报道基因构成的报道构件。一旦受体活化,g-蛋白受体激酶(grk)将细胞内特定残基磷酸化,并且所述诱导b-抑制蛋白2-tev蛋白酶的募集。tev蛋白酶识别和裂解tev位点,释放gal4-vp16融合蛋白,然后其转移至细胞核内。gal4-v16结合至uas元件,驱动b-内酰胺酶基因表达。用可渗透细胞的荧光底物ccf4-am检测b-内酰胺酶表达。所述底物由通过b-内酰胺环结合至荧光素的香豆素构成。无b-内酰胺酶时,具有405nm光的染料激发产生自香豆素至荧光素的fret,以及绿光(最大525nm)发射。底物的b-内酰胺酶裂解将香豆素荧光团自荧光素分离,并且405nm激发产生蓝色(最大460nm)发射。经蓝/绿发射比率检测测试。将oprδtangou2os细胞培养于生长培养基中(mccoy’s5a培养基,10%透析的fbs,非必需氨基酸,25mmhepes,1mm丙酮酸钠,青霉素/链霉素)。将二百万细胞添加至有30ml生产培养基的t175烧瓶中,并在37℃/5%co2下温孵4天,在所述时间点,它们是~70-90%融合的。经抽吸除去生长培养基,向烧瓶中加入5ml0.25%胰蛋白酶/edta,并轻轻洗涤细胞。然后,经抽吸除去胰蛋白酶。让细胞聚集,并通过轻敲烧瓶而脱离。将细胞悬浮于测试培养基中(高糖dmem,具有1%炭葡聚糖处理的fbs、非必需氨基酸、25mmhepes、1mm丙酮酸钠、青霉素/链霉素),研磨,计数,并通过离心而成丸。将细胞以160万细胞/ml再悬浮,并将10μl加入到黑色透明底的384-孔测试板(greinerpartnumber788092)的各孔中。将测试板置于增湿盒中,并在37℃/5%co2下孵育16-24小时。将溶解于dmso中的化合物依次用dmso在384-孔聚丙烯板中稀释。利用pintools,将50nl各化合物的稀释液加至测试板的各孔中。对照孔接受50nldmso。将板子放回培养器中,保持30分钟。用化合物预培养后,向测试板的化合物孔中加入50nl35umsnc80(激动剂激发)。对照孔中接受50nl2mm(100%响应对照)、50nl35umsnc80(ec80对照)或50nldmso(0%响应对照)。将测试板放回37℃/5%co2的培养器中,保持4小时。然后,将测试板从培养器中移出,并向各孔中加入2.5ulliveblazerccf4-am底物染料(invitrogen)。然后,将测试板在室温置于台子上,用铝箔覆盖,以避光,保持2小时。然后,将板子在荧光板读数器上,用405nm的激发波长,以及460nm和525nm的发射波长读数。利用蓝/绿发射比率计算结果。利用上文提到的等式计算抑制百分数,ic50是实现50%抑制需要的化合物浓度。在dormu下,下文表2中列出的代表性化合物都显示比3,000nm更大的ic50值。oprμ拮抗剂测试本测试的目的是确证合成的oprk1拮抗剂化合物的效力和选择性。本测试在β-抑制蛋白的膜募集中监测oprμ1活化。本测试利用β-半乳糖苷酶(β-gal)的低亲合力片段互补,监测gpcr-β-抑制蛋白的接近。本测试利用u20s细胞,其表达融合至互补β-gal片段(酶受体)的oprμ1。根据设计,作为拮抗剂的化合物将阻止受体活化,致使孔的荧光减少。将化合物一式三份测试,利用10-点、1:3稀释序列,从10微摩尔的标定浓度开始。将discoverxoprμ1-u20s细胞系在t175烧瓶中,于37℃、5%co2和95%相对湿度(rh)下常规培养。生长培养基由dmem/f121:1media构成,其补充有10%v/v热灭活胎牛血清、25mmhepes、非必需氨基酸、1mm丙酮酸钠、1x抗生素混合物(青霉素链霉素)+500ug/ml遗传霉素和300ug/ml潮霉素(选择抗生素)。在测试的第1天,将20ul测试缓冲液(discoverx’scellplatingreagent5)中的5000个细胞接种到384corning3570标准白色板的各孔中,并在37℃、5%co2和95%(rh)下温孵16-24小时。在第2天,将100nldmso中的测试化合物添加到适当的孔中,将100nldmso添加到对照孔中,并将板子在37℃、5%co2和95%(rh)下温孵30min。随后,将100nldamgooprμ1或dmso加到测试培养基(ec80激发由100nl50umdamgo构成,最终测试浓度=250nm,100%响应孔接受100nl200umdamgo)中。在37℃、5%co2和95%(rh)下温孵3小时后,将10ulpathhunterdetectionmix加入到各孔中,将板子置于板子旋转器/混合器上,保持~10分钟,然后在室温于黑暗中温孵1小时。在perkinelmer’senvision上测量孔发光。如下根据中值比率,计算抑制百分数:其中:化合物孔定义为含测试化合物的孔;ec80对照孔定义为含damgo激发(最终250nm)的孔=0%抑制;并且0%响应孔定义为含dmso的孔=100%抑制。ic50定义为实现50%抑制需要的化合物浓度。下表2中提供表示为代表性化合物抗κ阿片受体(kor)和μ阿片受体(mor)的ic50的活性。对于kor活性:“++++”指低于1nm的ic50;“+++”指1nm至低于10nm的ic50;“++”指10nm至低于100nm的ic50;并且“+”指100nm或更大的ic50。对于mor活性:“++++”指低于1nm的ic50;“+++”指1nm至低于10nm的ic50;“++”指10nm至低于100nm的ic50;并且“+”指100nm至比1,000nm更小的ic50;并且“-”指大于1,000nm的ic50。表2中还显示代表性化合物的κ特异性(即moric50/koric50)。如下表示选择性范围:++++指比μ阿片受体(mor)高1,000-倍的选择性,+++指100-至1,000-倍的选择性,++指10-至1,00-倍的选择性,并且+指低于10倍的选择性。表2代表性化合物的活性cpd.no.=化合物编号强啡肽a-诱导的β-抑制蛋白通路的活化还利用β-抑制蛋白测试(discoverx,fremontca),评价化合物142在oprk1上偏移的信号传导。在本测试中,将细胞用化合物142(5.1μm)预温孵,随后利用ec80浓度的强啡肽a(0.0498μm)激动剂激发(重复进行五次)。化合物142阻断强啡肽a-诱导的β-抑制蛋白通路的活化(ic50=3.1nm),提示为平衡的拮抗剂性质,并拮抗强啡肽a-诱导的受体内化(ic50=6.1nm)。图7。放射配体结合测试在利用[3h]二丙诺菲(diprenorphine)(600pm)的放射配体结合测试中,化合物142显示对人阿片受体κ1的有效抑制(hoprk1;ki=1.5nm),并显示高于人阿片受体μ1(hoprm1;ki=0.45μm)>300-倍的选择性(一式三份测试,n=3)。平均值±sem.图8实施例b-2体内活性小鼠甩尾研究利用甩尾测试,在小鼠中测定化合物142全身施用在阻断(-)-u-50,488-诱导的急性镇痛作用中的功效。简言之,测试小鼠基线反应,以测定它们对50℃热水浸没的甩尾的潜伏期(用15秒截断,以防止组织损伤)。oprk1激动剂(-)-u-50,488(15mg/kg,i.p.)、测试化合物(1mg/kg,i.p.)或者溶媒(10%dmso/10%tween80/80%盐水,5ml/kg,i.p.)施用1或24h以后,将(-)-u-50,488(15mg/kg,i.p.)施用至所有小鼠。将不同组小鼠在不同的预处理时间点处理。在(-)-u-50,488施用30min后,再评价甩尾潜伏期。在本项研究中,(-)-u-50,488在小鼠中产生强的镇痛作用,(-)-u-50,488使用前1h而不是24h施用的化合物142阻断该镇痛作用。当在无(-)-u-50,488施用时,化合物142没有作用。在不同群小鼠中,将成年、雄性、cd-1小鼠施用化合物142(30mg/kg,p.o.)或溶媒(施用10%dmso/10%tween80/80%盐水,随后1或24小时后施用(-)-u-50,488(20mg/kg,i.p.,生理盐水中)。测量从50℃水浴抽出其尾巴的潜伏期。在本项研究中,小鼠甩尾测试中,(-)-u-50,488使用前1h而不是24h施用的化合物142(30mg/kg,p.o.)阻断(-)-u-50,488诱导的镇痛作用,显示oprk1拮抗剂可逆的体内作用。图9小鼠催乳素研究将κ阿片拮抗剂以0.5mg/ml的浓度溶解于水中。将8-10周龄雄性c57bl/6j小鼠腹膜内使用10mpkκ阿片拮抗剂或溶媒水而进行处(treat),持续1小时。将u-69593κ阿片激动剂(sigma)以10mg/ml浓度溶解于45%2-羟基丙基-环糊精中。拮抗剂处理1小时后,将0.3mg/kgu-69593或溶媒0.3%2-羟基丙基-环糊精在颈背部皮下施用。激动剂处理30分钟后,将小鼠用co2安乐处死,并完成心脏穿刺。采集血液,并将血液置于k2edta包被的微量采血管中,并保持寒冷。将血在4℃、1,000xg旋转10分钟。除去血浆,并利用milliplexmousepituitarymagneticbeadpanel(mptmag-49k),将10ul用于测定催乳素水平。利用milliplexanalyzerluminex200,从各一式两份样品采集50个催乳素分析样品,并利用milliplexanalyst5.1软件定量测定催乳素的pg/ml。利用oprk1激动剂-诱导的催乳素激发方法,在8-10周龄雄性c57bl/6j小鼠中(n=3-4/组),测试化合物142的体内拮抗剂性质。尤其是,将化合物142(0.01-3.0mg/kg,ip;水[溶媒];1hptt)或化合物142(3,10或30mg/kg,po;水[溶媒];2hptt)施用至小鼠,随后注射oprk1-激动剂u69593(0.3mg/kg,sc;5%环糊精[溶媒];0.5hptt),并在终末操作0.5h后,通过心脏穿刺采集血浆样品。在所述研究中,u69593显著升高血浆催乳素浓度(p≤0.05vs溶媒),并且化合物142以剂量相关方式抑制催乳素的增加(p≤0.05vsu69593单独)。如图1所示,腹膜内注射(ip)后,用最小有效剂量(med)≤0.01mg/kg(参见图1a),化合物142有效阻断u69593-诱导的催乳素增加;并且口服(po)施用后,10mg/kg的化合物142显著抑制激动剂诱导的催乳素增加(参见图1b)。用相同方式测试其他化合物。表3显示所选化合物口服给药对血浆催乳素水平的作用结果。与u69593刺激和溶媒对照测量相比,基于催乳素水平的降低,评价结果:na表示无显著降低,+表示部分降低(<50%),++表示降低(>50%),+++表示降低至溶媒对照水平。图2显示化合物141ip注射(图2a)或po施用(图2b)后的结果。图3显示化合物145ip注射(图3a)或po施用(图3b)后的结果。图4显示化合物146ip注射(图4a)或po施用(图4b)后的结果。图5显示化合物147ip注射(图5a)或po施用(图5b)后的结果。图6显示化合物a的酒石酸盐ip注射后的结果。表3.口服催乳素数据(小鼠)化合物#1mg/kg3mg/kg10mg/kg30mg/kg时间34+2hr109nanana2hr110nanana2hr134+++++2hr136++++++2hr141++++++2hr142++++++++2hr145++++2hr146+++++++2hr147nana++2hr175+na++1hr176nana+1hr181++++++1hr182+++++1hr183na+++1hr196++1hr197++na++2hr214na+++1hr215++++1hr219+++++++++1hr220na++1hr250nanana2hr251na+++2hrna:无显著降低大鼠催乳素研究利用oprk1激动剂-诱导的催乳素激发方法,测试化合物142的体内拮抗剂性质。在本项研究中,将化合物142(10mg/kg和30mg/kg,po;水[溶媒];1hptt)施用至雄性sprague-dawley大鼠;n=8-10)后,注射oprk1-激动剂螺朵林(0.32mg/kg,sc;5%环糊精[溶媒];0.08hptt)。口服给药(b1)前5分钟和螺朵林给药(b2)前5分钟,然后螺朵林施用后5、30和60分钟通过尾静脉插管采集血液样品。在所述研究中,螺朵林显著升高血浆催乳素浓度,并且在5和60分钟时间点,用medí10mg/kg,化合物142抑制催乳素的升高。作为比较者,包括oprk1拮抗剂ly-2345302(10mg/kg,po;1hptt),并且在5、30和60分钟时间点,其抑制螺朵林-诱导的催乳素增加。图10大鼠应激-诱导的皮肤痛觉超敏(偏头痛的动物模型)在舒马普坦-诱导的潜在敏感大鼠中,评价全身施用后化合物42能阻断亮光应激(bls)-诱导的眼周和后爪痛觉超敏的功效。在本模型中,用微渗泵将大鼠输注舒马普坦(0.6mg/kg/天,s.c.),持续7天。在舒马普坦输注后第20和21天(d),每天将bls施用1小时,持续连续两天。在各bls期间之前的30min,施用化合物142(1mg/kg,i.p.)或溶媒(10%dmso/10%tween80/80%盐水,5ml/kg,i.p.)。在化合物142第二次施用之前d21,评价基线眼周和后爪的触觉阈值。计算原始数据和时间效应曲线上面积(areaoverthetimeeffectcurve)(aoc,从基线和每小时触觉阈值计算的)。各bls之前30min施用的化合物142(1mg/kg,i.p.)阻断舒马普坦-预处理大鼠中痛觉超敏的形成。用类似方式测试化合物34(5mg/kg,p.o.)和化合物196(5mg/kg,p.o.),并且其阻断舒马普坦-预处理大鼠中痛觉超敏的形成。脑透过研究利用体内微量透析方法,评价化合物142的脑透过性质,其中将大鼠在纹状体和颈静脉插管,并每隔30分钟采集样品。化合物142的纹状体暴露比相同动物收集的颈静脉样品中观察到的暴露高约4倍。外科手术:将成年、雄性spraguedawley大鼠麻醉,并植入颈静脉(jv)metaquant(mq)探针(3mm膜)。然后,将动物置于立体定位架上,并向纹状体插入第二个mq微透析探针(3mm膜):(ap)=距离前卤点+0.9mm,侧面(l)=距离中线+3.0mm和腹侧(v)=距离硬脑膜-6.5mm。手术后1天进行研究。样品收集和分析:用人造csf以0.15μl/min的流速灌输探针。每30分钟收集样品,并利用lc/ms分析。在brainson-line,llc(brisbane,ca)上进行试验。图11全细胞切片电生理学研究在大鼠中,通过g蛋白偶联内向整流k+通道(girks)的活化,kor选择性激动剂u69593引起一亚组腹侧被盖区多巴胺(vtada)神经元(但是不是非-da神经元)中突触后超极化。所述u69593作用的ec50是42nm。利用含有雄性、sprague-dawley大鼠vta的水平脑切片(~150μm),在原生组织中评价化合物142、化合物a、pf-04455242和ly2456302的功效、选择性和可逆性。图12。在本项研究中,在33℃,用2.5-4mω移液管,以电压钳制模式(vm=-60mv),进行全细胞记录,以保证放电率和/或膜电位测量。图13。不同浓度的化合物142、化合物a、pf-04455242和ly2456302(0.1-100nm)在细胞中经槽液施用(bathapplication),随后通过置于记录位置300μm内的加压喷射器递送的oprk1激动剂u69593(1μm)施用后,得到剂量-响应数据。每次施用由60秒激动剂的喷射,随后30秒对照人工脑脊液(acsf)洗涤构成。将数据报道为各响应细胞中各拮抗剂产生的u69593-诱导的外向电流抑制百分数。在对照试验中,将u69593重复施用至vta神经元产生类似的响应幅度。在探针试验中,将各细胞用至少一次u69593基线施用校准,并且将显示锁时应答(time-lockedresponse)的细胞用于拮抗剂表征。在本项研究中,化合物142显示完全拮抗性质和1.3nm的ic50。10nm和100nm的化合物142完全阻断u69593响应。所述非常类似于表达大鼠kors的异源性系统中的有效浓度,其中对于阻断(-)-u-50,488(3nm)的腺苷环化酶抑制,化合物142具有3.2nm的ic50。图14。相关化合物—化合物a—显示4.6nm的ic50。100nm剂量的化合物a几乎完全阻断u69593响应。图14b。还在本项测试中评价已知的kor拮抗剂pf-04455242。令人吃惊地,pf-04455242仅部分阻断u69593响应。用100nm和1μmpf-04455242,观察到最大60%的u69593响应阻断,具有19.6nm的绝对ic50。相对ic50是4.3nm。图14c。还测定ly2456302的剂量响应。对于ly2456302,发现0.3nm的绝对ic50,但是剂量响应形式是不正常的,不被hill等式很好-拟合。图14d。这些试验的另一个令人吃惊的特征是在ly2456302的100nm和1μm剂量,一亚组细胞响应u69593,具有内向电流。在中间前额皮质vtada神经元中,化合物142有效阻断kor活性。还测量化合物142特异性阻断计划到中间前额皮质(mpfc)的vtada神经元内u69593-诱导的响应的能力。在mpfc至vta的逆行标记神经元中特异性进行全细胞记录之前7天,将逆行示踪剂dii注射到mpfc神经元。在这些选择的神经元中,化合物142的ic50是2.2nm,在非选择神经元中的ic50的差异内。图15。与ly2456302相比,化合物142的kor选择性为了评价相对于mor和dor,化合物142和ly2456302对kor的选择性,测试vta神经元中所述两种受体类型上阻断选择性激动剂-诱导的响应的能力。将mor选择性激动剂damgo或者dor选择性激动剂dpdpe加压喷射到vta神经元,并在响应神经元中,将10nm各拮抗剂经槽液施用至少4分钟后,将激动剂再施用,该剂量的化合物142完全阻断用u69593的kor响应,其不影响对damgo或dpdpe的响应。相比之下,10nmly2456302始终如一地降低对damgo的响应。图16.急性洗出(acutewashout)norbni是最广泛使用的临床前kor拮抗剂工具。对于许多试验范例,norbni的一个主要缺点是甚至单次给药后,其持久生物活性的证据,而仪器上无可测量量的化合物。短效(可逆的)、选择性拮抗剂不仅可用于临床研发,而且可用于需要在数小时内通过(clear)大脑的配体的试验设计。在本项研究中,测试kor拮抗剂在vta脑切片中是否快速地从kors解离,足以在全细胞记录中观察洗出。在各试验中,测量u69593基线响应,然后将拮抗剂施用至切片,保持5-10分钟。根据剂量响应试验中观察的,所述间隔足够完全阻断u69593响应。然后,拮抗剂洗出开始后10分钟和/或20分钟,探测u69593响应。如预期的,norbni经典的离体剂量显示在20分钟无洗出(u69593基线响应的-27.13±17.41%,n=4)。另一方面,足够完全阻断u69593作用的10nm剂量的化合物142显示10min内的完全洗出。有趣地,化合物a不显示显著逆转,具有至多20分钟的洗出。所述数据表明相同化学系列中,在化合物的可逆作用方面可存在差异。pf-04455242显示一些洗出,20min但是不是10min后,u69593响应恢复。ly2456302在10或20min不显示实质上的逆转。图17。来自用其他kor拮抗剂的这些研究的结果表明pf-04455242显示部分拮抗剂活性,并且还在一亚组神经元中通过未知受体产生外向电流。切片试验期间,化合物a不显示kor阻断的洗出。此外,ly2456302可具有多于一个的结合位点,并且在存在100nm和1μm时,一亚组细胞对u69593响应,具有内向电流,表明这不是中性拮抗剂。所述数据提供电生理学证据,即化合物142是神经元中有效的、选择性的且短效的kor拮抗剂,所述神经元调节在神经行为病症中通常失调的脑回路。可将上述各种实施方案组合,以提供其他实施方案。将在本说明书中提到的和/或本申请数据页中列出的所有美国专利、美国专利申请公开书、美国专利申请书、外国专利、外国专利申请和非专利公开书以其全部引入文中作为参考。如果需要使用各种专利、申请和公开书的概念,以便还提供其他的实施方案,可修改实施方案的各方面。根据上文详细的描述,可对实施方案进行所述和其他变化。一般来讲,在下文权利要求书中,不应将所用的术语理解为限制权利要求至本说明书和权利要求书中公开的具体实施方案,而是应理解为包括所述权利要求赋予的所有可能的实施方案,以及全部的等效范围。因此,权利要求书不被公开书限制。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1