新型水溶性单支化和双支化的络合剂及相应的镧系元素络合物的制作方法
本发明涉及水溶性络合剂或配体、从这些络合剂获得的镧系元素络合物以及这些镧系元素络合物用于对分子进行标记并通过时间分辨荧光技术对它们进行检测的用途。
背景技术:
在过去20年中,镧系元素络合物的应用在生命科学领域中得到了极大的发展。其原因是这些荧光化合物具有有利的光谱特性,这使得它们成为用于对生物分子进行检测的首选标记。这些荧光化合物特别适合与相容的荧光团一起用于进行
已经描述了许多镧系元素络合物。例如latva等人公开了41个eu(iii)和tb(iii)络合物并研究了它们的发光(journalofluminescence1997,75,149)。特别地,化合物39由1,4,7-三氮杂环壬烷环(以下称为“tacn”)组成,其氮原子被衍生自苯乙炔基吡啶的发色团(chromophore/chrom)取代。尽管作者认为由该发色团和eu(iii)组成的络合物的量子产率良好,但是该络合物不适合与生物分子偶联。此外,由于该化合物是非常疏水的,其在水性介质中的使用可能存在问题。最后,该络合物的吸收在315nm处是最理想的,然而在用于生物测定的盘式分析仪中经常使用的激发波长更确切地是337nm。
d'aléo等人已经描述了由衍生自二吡啶羧酸的三个配体组成的镧系元素络合物的合成(inorganicchemistry2008,47,10258)。这些配体中的一种(l1)由被苯乙炔基基团取代的二吡啶羧酸分子组成,其本身在苯基基团上携带聚乙二醇醚-氧化物(以下称为“peg”)。根据作者所述,peg基团赋予了该产品在水性介质和有机溶剂中的良好溶解性。然而,这些络合物在水性介质中不够稳定并且不能用于生物缀合反应。
已公开了几种其它镧系元素络合物并且一些已被商业化开发:可以特别提及的是大的多环镧系元素穴状化合物(ep-a-0180492、ep-a-0321353、ep-a-0601113、wo2001/96877;wo2008/063721);包含与二乙撑三胺五酸单元连接的、衍生自香豆素的单元的镧系元素络合物(us5,622,821);以及包含吡啶衍生物(us4,920,195、us4,761,481)、联吡啶衍生物(us5,216,134)或三联吡啶衍生物(us4,859,777、us5,202,423、us5,324,825)的那些镧系元素络合物。
专利申请wo2013/011236描述了下式的络合剂:
在该申请中,发明人设想了具有三个发色团的络合物以增加亮度。另外,为了使这些络合物具有水溶性,发明人使用了peg基团。尽管这些peg基团从“总电荷”的观点来看是中性的,但它们赋予络合物在塑料和玻璃上的吸附性质,这使得它们难以用于免疫测定。
专利申请wo2014/111661描述了包含三个发色团基团的络合剂,由下式表示:
在该申请中,发明人用阴离子或阳离子带电基团代替peg基团。这些络合物是完全可溶的,不再吸附在塑料或玻璃上。这些络合物具有相当大的星形结构和非常高的亮度性质。
专利申请wo2014/162105涉及在有机部分上含有至少两个甜菜碱基团的镧系元素络合物,赋予它们在水和生物介质中溶解度方面的有利性质。这些增溶基团表现为限制活细胞的非特异性吸附现象。另一方面,这些络合物具有三个相同或不同的发色团,如在申请wo2014/111661中,它们产生相当大的星形结构和良好的亮度性质。
专利申请wo2014/147288和wo2016/066641描述了具有高摩尔吸收系数的tacn络合物。这些络合物具有高亮度,但也具有三个发色团。发明人正在寻找最亮的可能络合物以便具有最佳的能量转移(fret)。络合物的星形结构再次是大体积的,并且它们的亮度高。
由于发色团是由冗长且乏味的合成得到的合成子,在三氮杂环壬烷大环上引入三个发色团并不总是有利的。tacn络合物中三个发色团的存在增加了分子的大小,这增加了生物分子的空间位阻。然而,发色团的数目越高,亮度越高,因为该参数取决于分子的量子产率和摩尔吸收系数(ε(epsilon))。通过向分子中引入三个发色团,增加了亮度。
技术实现要素:
在本发明中,我们已经发现,在基于时间分辨fret的免疫测定中,络合物的亮度与其检测生物靶标的能力之间没有相关性,在所述时间分辨fret中,将络合物用作能量供体。本发明旨在克服现有技术的缺点,即,通过引入一个或两个发色团来简化合成,以减少空间位阻,并最终获得在能量转移方面具有至少相同性能的荧光探针。
因此,本发明旨在提供荧光镧系元素络合物,当在约337nm下进行激发时,该荧光镧系元素络合物具有比现有技术的化合物更低的亮度,但其在基于时间分辨fret的免疫测定中检测生物靶标的能力与携带三个发色团的类似物相当或更好。这些络合物还具有在水性介质中的良好溶解性、适用于fret实验的发射光谱以及由于它们的尺寸较小而用于标记生物分子的良好实用性。
附图说明
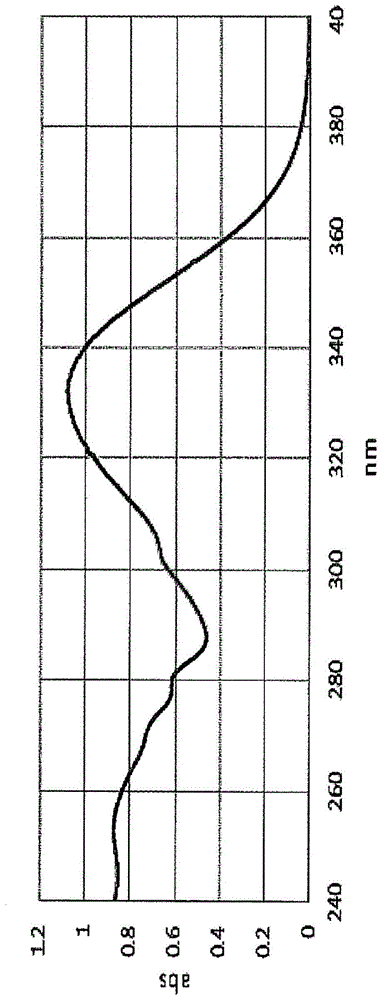
图1至图3分别表示代表本发明的络合物的uv光谱、色谱图和质谱。
图4示意性地表示用于测试代表本发明的络合物的有效性的免疫测定。
图5表示在实施图4中的免疫测定期间测量的fret信号(δf(deltaf))。
图6示意性地表示用于测试代表本发明的络合物的有效性的免疫测定。
图7表示在实施图6中的免疫测定期间测量的fret信号(δf)。
具体实施方式
络合剂
本发明所述的络合剂为式(i)的化合物:
其中:
-发色团1、发色团2和发色团3各自独立地表示下式的基团:
条件是,式(i)化合物(必须)包括:(i)选自基团(ia)和基团(ib)的一个或两个基团,以及(ii)至少一个l1-co-r基团;
-r为-or2或-nh-e基团;
-r1为-co2h或-p(o)(oh)r3基团;
-r2为h或(c1-c4)烷基;
-r3为(c1-c4)烷基、优选甲基;任选地由-so3-基团取代的苯基,所述-so3-基团优选在间位或对位;或苄基;
-l1为直接键合;-(ch2)r-基团,所述-(ch2)r-基团任选地由选自氧原子、氮原子和硫原子的至少一个原子中断;-ch=ch-基团;-ch=ch-ch2-基团;-ch2-ch=ch-基团;或peg基团;
-l2为二价连接基团;
-g为反应性基团;
-e为-ch2-(ch2)s-ch2-so3-或-(ch2)s-n+alk1alk2alk3基团,或磺基甜菜碱;
-r为1至6的整数,优选1至3;
-s为0、1或2;
-alk1、alk2、alk3可相同或不同,表示(c1-c6)烷基。
因此,由于式(i)的结构包含三个发色团(发色团1、发色团2和发色团3),本发明所述的络合剂包含选自基团(ia)和基团(ib)中的一个或两个基团,并因此包含选自基团(ic)和基团(id)中的一个或两个基团。对式(i)化合物的附加要求是结构中必须存在至少一个l1-co-r基团。
peg基团是指式-ch2-(ch2och2)y-ch2och3的聚乙二醇基团,其中y是1至5的整数。
磺基甜菜碱是指选自以下的基团:
r4表示(c1-c6)烷基,优选甲基或乙基,t等于1、2、3、4、5或6,并且优选等于1或2;式-(ch2)2n+(ch3)2-(ch2)3-so3-的磺基甜菜碱是优选的。
根据ph,-so3h、-co2h和-po(oh)2基团为去质子化形式或非去质子化形式。因此,这些基团在下文中还指基团-so3-、-co2-和-po(oh)o-,反之亦然。
第一优选的络合剂族由式(i)的化合物组成,其中发色团1是式(ia)的基团,发色团2和发色团3各自是相同或不同的式(ic)的基团。在一个实施方式中,发色团2和发色团3相同。在可与前述实施方式组合的另一个实施方式中,r1是-co2h或-p(o)(oh)r3基团,其中r3是(c1-c4)烷基或苯基。
第二优选的络合剂族由式(i)的化合物组成,其中发色团1和发色团2各自是相同或不同的式(ib)的基团,并且发色团3是式(id)的基团。在一个实施方式中,发色团1和发色团2相同。在可与前述实施方式组合的另一个实施方式中,r1是-co2h或-p(o)(oh)r3基团,其中r3是(c1-c4)烷基或苯基。
在这两个优选的族中,优选的亚族是其中络合剂包括一个或多个以下特征的那些:
-r2为h;
-l1为直接键合;-(ch2)r-基团,其任选地由选自氧原子和硫原子的至少一个原子中断,并且r=2或3;-ch=ch-基团;-ch=ch-ch2-基团;或-ch2-ch=ch-基团;
-e为-ch2-(ch2)s-ch2-so3-基团,s=0或1;-(ch2)s-n+alk1alk2alk3,表示(c1-c4)烷基的alk1、alk2、alk3相同或不同,并且s=0或1;或者下式的基团:
其中r4是(c1-c4)烷基并且t是1或2。
在本发明的一个实施方式中,当式(i)的络合剂包含几个e基团时,这些基团中的至多一个表示磺基甜菜碱。
由间隔臂l1或l2携带的反应性基团g允许使本发明所述的化合物偶联待产生荧光的物质(例如有机分子、肽或蛋白质)。用于使两个有机分子缀合的技术基于反应性基团的使用,并且是本领域技术人员的常识的一部分。这些经典技术描述于例如bioconjugatetechniques,g.t.hermanson,academicpress,2008年第二版,p.169-211。
典型地,反应性基团是亲电基团或亲核基团,当其分别在合适的亲核或亲电基团存在下时可以形成共价键。包含反应性基团的本发明所述的化合物与携带官能团的有机分子、肽或蛋白质之间的缀合反应使得包含反应性基团的一个或多个原子的共价键的形成。
优选地,反应性基团g是衍生自以下化合物中的一个的基团:丙烯酰胺、活化胺(例如,尸胺或乙二胺)、活化酯、醛、卤代烷、酸酐、苯胺、叠氮化物、氮杂环丙烷、羧酸、重氮烷、卤代乙酰胺、卤代三嗪(例如一氯三嗪、二氯三嗪)、肼(包括酰肼)、亚氨酸酯、异氰酸酯、异硫氰酸酯、马来酰亚胺、磺酰卤、硫醇、酮、胺、酰卤、琥珀酰亚胺酯、羟基琥珀酰亚胺酯、羟基磺基琥珀酰亚胺酯、叠氮基硝基苯基、叠氮基苯基、3-(2-吡啶基二硫代)-丙酰胺基、乙二醛、三嗪、乙炔基团,以及特别是选自下式基团的基团:
其中w从0至8变化,v等于0或1,ar是包含1至3个杂原子的饱和或不饱和的5元或6元杂环,所述5元或6元杂环任选地由卤素原子取代。
优选地,反应性基团g是胺(任选地以-nhboc形式保护)、琥珀酰亚胺酯、卤代乙酰胺、肼、异硫氰酸酯、马来酰亚胺基团或羧酸(任选地以-co2me、-co2tbu基团保护)。在后一种情况下,为了与亲核物质反应,酸必须活化成酯。
反应性基团g通过间隔臂l1或l2结合到络合剂上,所述间隔臂l1或l2有利地由二价有机基团构成。特别地,间隔臂l2可以选自:
-直接键合;
-直链或支链的c1-c20、优选c1-c8亚烷基基团,所述亚烷基基团任选地含有一个或多个双键或三键;
-c5-c8环亚烷基基团;c6-c14亚芳基基团;
所述亚烷基、环亚烷基或亚芳基基团任选地含有诸如氧、氮、硫、磷的一个或多个杂原子,或一个或多个氨基甲酰基或甲酰胺基基团,并且所述亚烷基、环亚烷基或亚芳基基团任选地由1至5(优选1至3)个c1-c8烷基、c6-c14芳基、磺酸基或氧代基团取代;
-选自下式的二价基团的基团:
-(ch2)n-、-(ch2)n-o-(ch2)m-o-(ch2)p-、
-(ch2)n-nh-、-(ch2)n-nh-(ch2)m-、
其中n、m、p、q是1至16的整数,优选1至5,e是1至6的整数,优选1至4。
在式(ia)的基团中,-l2-g基团优选由反应性基团g和间隔臂l2组成,所述反应性基团g选自羧酸(任选地以-co2me、-co2tbu基团的形式保护)、胺(任选地以-nhboc形式保护)、琥珀酰亚胺酯、卤代乙酰胺、肼、异硫氰酸酯、马来酰亚胺基团,所述间隔臂l2由含有1至5个碳原子的亚烷基链或选自下式基团的基团组成:
其中,n、m是1至16的整数,优选1至5,e是1至6的整数,优选1至4,基团g连接至这些二价基团的任一端。
络合物
本发明还涉及由上述络合剂络合的镧系元素原子组成的镧系元素络合物,所述镧系元素选自:eu3+、sm3+、tb3+、gd3+、dy3+、nd3+、er3+。镧系元素优选为tb3+或eu3+,甚至更优选eu3+。
通过使本发明所述的络合剂与镧系元素盐反应来制备此类络合物。因此,在溶剂(与这些盐相容的乙腈、甲醇或其它溶剂)或缓冲液中,在搅拌几分钟后,1当量络合剂与1至5当量镧系元素盐(处于氯化物、乙酸盐或三氟甲磺酸盐形式的铕或铽)之间的反应产生相应的络合物。
如前所述,所获得的荧光络合物具有优异的光物理性质,特别是在它们的量子产率、它们的发光寿命和它们的激发光谱方面,其特别适合于频闪灯或在约337nm下的激光激发。此外,它们的发射光谱的谱带分布集中在620nm左右,因此当将fret与青色素或别藻蓝蛋白型的受体(例如由cisbiobioassays出售的xl665)一起使用时,给予络合物杰出且非常有利的性质。与现有技术的带有三个发色团的络合物相比,由于这些络合物在含有大多数二价阳离子(ca2+、mg2+等)或edta的生物环境中的高稳定性,它们的发光保持优异。
缀合物
含有g基团的本发明所述的络合剂和镧系元素络合物特别适合用于对含有官能团的有机或生物分子进行标记,所述官能团能够与反应性基团反应以形成共价键。因此,本发明还涉及镧系元素络合物用于对感兴趣分子(蛋白质、抗体、酶、激素等)进行标记的用途。
本发明还涉及用本发明所述的络合物标记的分子。如果所有的有机或生物分子具有能够与反应性基团反应的官能团,则它们可与本发明所述的络合物缀合。特别地,本发明所述的缀合物包含本发明所述的络合物和选自以下的分子:氨基酸、肽、蛋白质、抗体、糖、糖链、核苷、核苷酸、寡核苷酸、酶底物(特别是自杀酶底物,例如苄基鸟嘌呤或苄基胞嘧啶(作为snaptag和cliptag出售的酶底物))、氯烷烃(以名称halotag出售的酶的底物)、辅酶a(以名称acptag或mcptag出售的酶的底物)。
合成
下面示意性地描述用于本发明所述的络合剂(配体)和络合物的制备的通常策略(方案1:单天线(singleantenna)和方案2:双天线(twoantennae)),并且在实验部分中进行更为详细地描述。
方案1
从boc-单保护的三氮杂环壬烷大环1引入两个吡啶单元,其将用于连接两个增溶基团。除去保护基团boc,然后将天线(发色团)添加到大环上,产生配体3。酯(羧酸酯和次膦酸酯)的水解采用碱性条件以常规方式进行。然后,这允许铕原子得以并入,从而形成络合物5(两种水溶性官能团从其中引入)。最后,在由天线(发色团)携带的保护基团boc脱保护之后,络合物被官能化(7),使得它们可以缀合在生物分子上。
方案2
通过使用类似的策略获得双天线系统,但反转吡啶基单元和发色团的引入顺序。此次,首先引入天线以产生化合物8。在boc基团去除后,引入最后的吡啶基单元。顺序是相同的,即酯官能团(羧酸酯和次膦酸酯)的水解,铕络合物的形成,两种水溶性官能团(此次这些官能团由发色团携带)的引入,然后引入产生双天线族13的官能团。
1)吡啶基模块(brick)的制备
以下方案(3-11)描述了三官能吡啶基衍生物的各种合成途径:
-在2位上,络合官能团(羧酸或次膦酸);
-在4位上,允许引入水溶性基团的官能团(甲酯官能团)或允许引入允许并入官能团的官能团(保护的胺官能团和叔丁酯官能团);
-以及最后,在6位上,甲醇官能团,其被转化为相应的甲磺酸酯,以便能够与tacn环的胺反应。
方案3
以上描述了合成子14a-14c的合成(参见申请wo2013/011236和wo2014/111661)。从这些合成子,通过一系列的三个反应获得一系列化合物17a-17f:heck反应用于在吡啶衍生物和烯烃之间产生碳-碳键。例如,在专利申请ep-a-2002836中描述了该过程。通过催化氢化还原双键,然后进行甲基化反应,得到化合物17a-17f。或者,保留双键以使体系难以弯曲(stiffen),并对水增溶基团施加顶端取向(18a-18f)。
方案4
通过按照相同的策略并使用相应的烯烃获得处于叔丁酯形式的化合物21a-21f和22a-22f(方案4)(系列17和18的类似物)。
方案5
使用相应的烯烃,按照相同的策略获得处于nhboc形式的化合物25a-25f和26a-26f(方案5)(系列17和18的类似物)。
方案6
根据类似的策略制备化合物28a-28c(无碳链)(25系列的类似物)。nhboc基团的引入例如使用在综述文章tetrahedronletters2010,51,4445中描述的方法进行。
方案7
根据方案7中描述的方法制备吡啶基衍生物,在所述吡啶基衍生物上在携带官能团(co2r或nhboc)的脂族接头和芳环(吡啶)之间的4位插入氧原子。将白屈氨酸29酯化为甲基二酯,然后使用mitsunobu反应引入携带官能团的接头(过程描述于例如organicbiomolecularchemistry,2012,10,9183)。使用硼氢化钠的单还原产生处于一元醇形式的化合物32a-32c,然后将其转化为相应的甲磺酸化衍生物33a-33c。
方案8
4位的甲酯官能团可以直接固定到芳环(吡啶)上。在这种情况下,从首先酯化的市售化合物34开始是有必要的。然后在m-cpba存在下氧化吡啶,得到相应的n-氧化物衍生物36。n-氧化物官能团容易与三氟乙酸酐反应,其在水解后在6位被重排为甲醇官能团。后者在常规条件下被甲磺酸化,得到化合物38。
方案9
使用化合物39制备类似的次膦酸酯衍生物44a-44b,所述化合物39首先被酯化,然后转化成次膦酸酯41a-41b。反应顺序的其余部分与用于合成化合物38的反应顺序相同。
方案10
根据方案10中描述的反应顺序制备衍生物51a-51b。在该实例中,使用硫代乙醇酸乙酯或硫代乙醇酸叔丁酯引入酯官能团。
方案11
根据方案11中描述的合成路线制备次膦酸酯类似物56a-56d。
2)发色团的制备
方案12
根据申请wo2013/011236和wo2014/111661中详述的方案制备发色团58a-58c(方案12)和60a-60c(方案13)。
方案13
3)单天线络合物的合成
方案14
从boc-单保护的tacn大环、吡啶衍生物(py)缩合生成化合物61a-61s。将大环脱保护,并在配体上引入相应的发色团(z与py携带的那些相同)。酯官能团被水解(系列63)并且铕在不同配体中络合以产生系列络合物64a-64s。
方案15
在系列64a-64s中,通过引入两个水溶性基团使化合物可溶于水性介质中:这些基团在性质上是阴离子型的(磺酸盐)或中性的(两性离子:磺基甜菜碱),或阳离子型的(季铵)。
方案16
最后,在三氟乙酸存在下消除boc基团,这生成nh2官能化的本发明的络合物(66)。
4)双天线络合物的合成
在方案17-方案20中描述了双天线络合物的合成。
方案17
合成开始于单保护的tacn与三种发色团(羧酸酯、次膦酸甲酯和次膦酸苯酯)的烷基化反应。除去保护基团boc,并在最后的tacn烷基化位点引入携带与发色团相同的z的相应吡啶,得到化合物67a-67af。
方案18
水解配体并将铕原子引入到大环中,得到系列68。
方案19
然后在两个发色团上引入水溶性基团(e1-e5)(方案19)。它们在性质上是阴离子型、中性或阳离子型。
方案20
最后,在三氟乙酸存在下除去boc或叔丁基酯基团,得到化合物70a-70af(方案20)。
实验部分
使用的缩写:
acoet:乙酸乙酯
acoh:乙酸
boc:叔丁氧羰基
tlc:薄层色谱
cdcl3:氘代氯仿
chcl3:氯仿
csco3:碳酸铯
cui:碘化铜(i)
ch2cl2/dcm:二氯甲烷
diad:偶氮二甲酸二异丙酯
dipea:二异丙基乙胺
dmf:二甲基甲酰胺
dmso:二甲基亚砜
et:乙基
et3n/tea:三乙胺
esi+:正模式电喷雾电离
etoh:乙醇
d:天
hatu:(邻-(7-氮杂苯并三唑-1-基)-n,n,n′,n′-四甲基脲六氟磷酸酯)
h2o:水
h2so4:硫酸
hno3:硝酸
hplc:高效液相色谱法
lc-ms:高效液相色谱-质谱联用
lioh/lithine:氢氧化锂
lncl3:氯化镧
m-cpba:间氯过苯甲酸
me:甲基
mecn:乙腈
meoh:甲醇
mgso4:硫酸镁
ms:甲磺酰基
mscl:甲磺酰氯
nacl:氯化钠
nah:氢化钠
pd(dppf)cl2:[双(二苯基膦基)二茂铁]二氯化钯(ii)
pd(pph3)4:四(三苯基膦)钯(0)
pd/c:钯炭
pd(oac)2:乙酸钯(ii)
ph:苯基
pph3:三苯基膦
ptf:熔点
py:吡啶
rf:溶剂前沿
rt:保留时间
rt:室温
tbu:叔丁基
tea:三乙胺
tfa:三氟乙酸
thf:四氢呋喃
tms:三甲基硅烷基
ts:对甲苯磺酰基
tstu:邻-(n-琥珀酰亚胺基)-1,1,3,3-四甲基脲四氟硼酸酯
uplc-ms:超高效液相色谱-质谱联用
xphos:2-二环己基膦-2′,4′,6′-三异丙基联苯。
色谱
在铝箔上的merck60f254硅胶板或在铝箔上的merck60f254中性氧化铝板(e型)上进行薄层色谱。
在两个装置上进行分析型和制备型高效液相色谱(hplc):
-分析型hplc:thermoscientific,四元泵p4000,具有氘灯(190nm-350nm)的uv检测器1000,分析柱watersxbridgec18,3.5μm,4.6×100mm。
-制备型hplc:shimadzu,2个lc-8a泵,varianprostar二极管阵列uv检测器,watersxbridge制备柱c18,5μm:19×100mm或50×150mm。
分析用超高效液相色谱(uplc)在watersacquityhclass装置上进行,该装置具有pda类型的uv二极管条带检测器或sqd2型的简单四极质量检测器作为检测器。所使用的探针是正模式的电喷雾:3.2kv的毛细管电压~30v的锥孔电压(conevoltage)。
在merck60硅胶(0.040mm-0.063mm)上进行硅胶柱层析。氧化铝柱层析在sigma-aldrich氧化铝(中性,活化,brochmanni)上进行。
梯度a
watersacquityc18柱,
梯度b
watersxbridgec18柱,5μm,50×150mm-a/水25mmteaacph7b/乙腈,t=0min,10%b-t=19min,60%b-100ml/min。
梯度c
watersacquityc18柱,
梯度d
watersxbridgec18,5μm,20×100mm-a/水25mmteaacph7b/乙腈,t=0min,5%b-t=19min,60%b-20ml/min。
梯度e
watersxbridgec18,5μm,20×100mm-a/水25mmteaacph7b/乙腈,t=0min,2%b-t=19min,40%b-20ml/min。
梯度f
watersxbridgec18,5μm,20×100mm-a/水25mmteaacph6b/乙腈,t=0min,2%b-t=19min,40%b-20ml/min。
光谱
·核磁共振
使用配备有bbfo(多核5mm直径,梯度z和锁场2h测量探针)的brukeravance400mhznanobay光谱仪(9.4特斯拉磁体)进行nmr光谱(1h、13c和31p)。化学位移(δ)以百万分之一(ppm)表示。使用以下缩略词:
s:单峰;bs:宽单峰;d:二重峰;t:三重峰;q:四重峰;m:多重峰;dd:双二重峰;td:三重双峰;qd:四重双峰;ddd:双二倍二重峰。
·质谱(lrms)
使用配备有watersxbridgec18,3.5μm,4.6×100mm柱的具有esi/apci多模源的单四极waterszq2000光谱仪或sqd2型的单四极质谱仪进行(lc-ms)质谱。
·高分辨率质谱(hrms)
使用配备有气动辅助大气压电离源(api)的qstarelite质谱仪(appliedbiosystemssciex)进行分析。在以下条件下以正电喷雾模式对样品进行电离:电喷雾电压(isv):5500v;孔电压(orificevoltage)(or):20v;喷雾器气体压力(空气):20psi。用飞行时间(tof)分析仪获得高分辨率质谱(hrms)。以具有双重内部校准的三重复进行精确质量的测量。
实施例
化合物1:根据申请wo2013/011236和wo2014/111661中描述的过程制备化合物1。
化合物14a-14c:根据申请wo2013/011236和wo2014/111661中描述的过程制备化合物14a-14c。
化合物15a:在100ml施兰克(schlenk)烧瓶中,将化合物14a(440mg,1.5mmol)溶解在无水dmf(10ml)中以得到无色溶液。向反应混合物中一次性加入三(邻甲苯基)膦(91mg,0.3mmol)、pd(oac)2(33.7mg,0.15mmol)、tea(0.314ml,2.252mmol)和丙烯酸甲酯(0.203ml,2.252mmol)。反应在70℃下搅拌5小时。通过uplc-ms(梯度a)监测反应进程。在这段时间之后,反应完成。减压浓缩反应混合物,在acoet(50ml)中稀释,用水(2×50ml)洗涤,然后用nacl饱和水(50ml)洗涤。有机相用mgso4干燥,过滤并减压浓缩。使用dcm/meoh溶剂(梯度为100/0至99/1),通过硅胶柱层析法纯化粗产物,得到白色粉末形式的化合物15a(233mg,62%)。
pf=156.4-156.9℃–hplc梯度a-rt=2.03min–[m+h]+,m/z251.9–rf=0.41(硅胶,二氯甲烷–甲醇96:4)–hrms(esi+)计算值c12h14no5+[m+h]+,m/z252.0866,测量值:252.0868–1hnmr(400mhz,cdcl3)δ:8.13(s,1h,pyh5),7.67(d,j=16.2hz,1h),7.65(s,1h,pyh3),7.19(dd,j=;16.2hz,2h,hc=ch),6.71(d,j=16.2hz,1h),4.91(s,2h,ch2-oh),4.04(s,3h,py-coome),3.85(s,3h,coome),3.49(sl,1h,oh);13cnmr(100mhz,cdcl3)δ:166.20(coome),165.24(py-coome),161.59(pyc2),147.97(py-c=c),143.74(pyc6),140.92(pyc4),123.77(pyc3),122.08(pyc5),121.86(py-c=c),64.68(ch2-oh),53.10(py-co2ch3),52.19(co2ch3).
化合物15b-15f:使用相应的烯烃,根据与合成15a的过程相同的过程制备这些化合物。
化合物16a:在50ml烧瓶中,将化合物15a(233mg,0.927mmol)溶解在meoh(10ml)中以得到无色溶液。向反应混合物中一次性加入10%pd/c(23.69mg,0.022mmol)。将反应物在rt下在鼓泡的氢气下搅拌2小时。通过uplc-ms(梯度a)监测反应进程。在这段时间之后,反应完成。将反应混合物通过22μm尼龙过滤器过滤,蒸发至干,得到白色粉末形式的化合物16a(231mg,98%)。pf=133.2-136.4℃–hplc梯度a-rt=1.86min–[m+h]+,m/z253.2–hrms(esi+)计算值c12h16no5+[m+h]+,m/z254.1023,测量值:254.1024–1hnmr(400mhz,cdcl3)δ:7.9(s,1h,pyh5),7.41(s,1h,pyh3),4.85(s,2h,ch2-oh),4.01(s,3h,py-coome),3.69(s,3h,coome),3.05(t,j=7.6hz,2h,py-ch2-ch2),2.71(t,j=7.6hz,2h,py-ch2-ch2);13cnmr(100mhz,cdcl3)δ:172.41(coome),165.65(py-coome),160.49(pyc2),151.67(pyc4),147.22(pyc6),140.92(pyc3),123.96(pyc3),123.9(pyc5),64.62(ch2-oh),52.91(py-co2ch3),51.91(co2ch3),33.97(py-ch2-ch2),30.07(py-ch2-ch2)。
化合物16b-16f:根据与用于合成16a的过程相同的过程制备这些化合物。
化合物17a:在100ml烧瓶中,将化合物16a(231mg,0.912mmol)溶解在无水thf(30ml)中以得到无色溶液。将tea(0.127ml,0.912mmol)和mscl(72μl,0.912mmol)一次性加入置于冰浴中的反应混合物中。使混合物升温至rt并搅拌15分钟。通过uplc-ms(梯度a)监测反应进程。在这段时间之后,反应完成。减压浓缩反应混合物,在dcm(50ml)中稀释,用水(2×25ml)洗涤,然后用nacl饱和水(20ml)洗涤。有机相用mgso4干燥,过滤并减压浓缩,得到白色粉末形式的化合物17a(249mg,82%)。hplc梯度a-rt=3.2min-[m+h]+,m/z332.3–rf=0.23(硅胶,二氯甲烷–甲醇98:2)–hrms(esi+)计算值c13h18no7s+[m+h]+,m/z332.0799,测量值:332.0799-1hnmr(400mhz,cdcl3)δ:7.98(s,1h,pyh5),7.54(s,1h,pyh3),5.41(s,2h,ch2-oms),4.00(s,3h,py-coome),3.69(s,3h,coome),3.16(s,3h,oms),3.07(t,j=7.5hz,2h,py-ch2-ch2),2.72(t,j=7.5hz,2h,py-ch2-ch2);13cnmr(100mhz,cdcl3)δ:172.23(coome),165.27(py-coome),154.56(pyc2),152.46(pyc4),147.93(pyc6),125.19(pyc3),125.03(pyc5),70.97(ch2-oms),53.08(py-co2ch3),51.94(co2ch3),38.05(ch2-oso2ch3),33.87(py-ch2-ch2),30.07(py-ch2-ch2).
化合物17b-17f:根据与用于合成16a的过程相同的过程制备这些化合物。
化合物18a-18f:根据与用于合成17a的过程相同的过程制备这些化合物。
化合物19a-19f:根据与用于合成15a的过程相同的过程制备这些化合物。
化合物20a-20f:根据与用于合成16a的过程相同的过程制备这些化合物。
化合物21a-21f:根据与用于合成17a的过程相同的过程制备这些化合物。
化合物22a-22f:根据与用于合成17a的过程相同的过程制备这些化合物。
化合物23a-23f:使用相应的烯烃,根据与用于合成15a的过程相同的过程制备这些化合物。
化合物24a-24f:根据与用于合成16a的过程相同的过程制备这些化合物。
化合物25a-25f:根据与用于合成17a的过程相同的过程制备这些化合物。
化合物26a-26f:根据与用于合成17a的过程相同的过程制备这些化合物。
化合物27a-27c:根据文章:tetrahedronletters2010,51,4445中描述的过程制备化合物27a-27c。
化合物28a-28c:根据与用于合成17a的过程相同的过程制备这些化合物。
化合物29:该化合物可商业化获得。
化合物30:根据文章:daltontransactions2010,39,707中描述的过程制备化合物30。
化合物31a-31c:根据文章:organicbiomolecularchemistry2012,10,9183中描述的过程制备化合物31a-31c。
化合物32a-32c:根据文章:journaloforganicchemistry2010,75,7175中描述的过程制备化合物32a-32c。
化合物33a-33c:根据文章:journaloforganicchemistry2010,75,7175中描述的过程制备化合物33a-33c。
化合物34:该化合物可商业化获得。
化合物35:根据文章:bioorganicchemistry2014,57,148中描述的过程制备化合物35。
化合物36:根据文章:carbohydrateresearch2013,372,35中描述的过程制备化合物36。
化合物37:根据wo2014/111661中描述的过程制备化合物37。
化合物38:根据与用于合成17a的过程相同的过程制备该化合物。
化合物39:这一化合物可商业化获得。
化合物40:根据文章:bioorganicchemistry2014,57,148中描述的过程制备化合物40。
化合物41a-41b:使用相应的催化剂,根据申请wo2014/111661中描述的过程制备化合物41a-41b。
化合物42a-42b:根据与用于合成36的过程相同的过程制备化合物42a-42b。
化合物43a-43b:根据与用于合成37的过程相同的过程制备化合物43a-43b。
化合物44a-44b:根据与用于合成17a的过程相同的过程制备化合物44a-44b。
化合物45:可商业化获得。
化合物46:根据文章:chemistry–aeuropeanjournal,2014,20,3610中描述的过程制备化合物46。
化合物47:在rt下将化合物46(0.313g,2.04mmol)溶解在h2so4(11ml)中,然后将溶液在冰浴中冷却。向该混合物中滴加hno3(9.7ml),并将溶液加热至100℃持续2天。将混合物冷却至rt,然后倒入碎冰(100g)中。1小时后,用ch2cl2(3×50ml)萃取水相,合并有机相,用mgso4干燥,粗产物通过硅胶柱层析法用溶剂混合物(ch2cl2-acoh,98/2)纯化,得到白色固体(224mg,56%)。rf(ch2cl2/acoh,98/2)=0.38;ptf:147℃;1hnmr(400mhz,cdcl3,δ):16.49(s,1h,cooh),9.08(s,1h,h3),8.36(s,1h,h5),2.75(s,3h,py-ch3);13cnmr(101mhz,cdcl3,δ):159.4(cooh),152.4(c6),144.4(c4),138.7(c2),123.1(c5),121.7(c3),18.4(py-ch3);ms计算值c7h7n2o5199.036。测量值199.035[m+h]+。
化合物48:在rt下将化合物47(2.9g,14.7mmol)溶于无水meoh(3ml)中。向该溶液中滴加h2so4(200μl),并将溶液加热至65℃持续3天。将溶液冷却至rt并在减压下除去溶剂。向残余物中加入h2o(30ml)并用acoet(3×20ml)萃取溶液。合并有机相,用5%碳酸氢钠溶液(2×20ml)洗涤,然后用饱和盐水溶液(20ml)洗涤。在经mgso4干燥后,过滤、减压除去溶剂,得到化合物48,其用于随后的合成而无需进一步纯化(57mg,76%)。1hnmr(400mhz,cdcl3,δ):8.33(d,1h,4j3.1,h5),8.19(d,1h,4j3.1,h3),4.02(s,3h,ch3co),2.57(s,3h,py-ch3);13cnmr(100mhz,cdcl3,δ):160.8(coome),152.7(c6),142.1(c4),140.5(c2),121.4(c5),119.3(c3),53.8(och3),18.3(py-ch3);ms计算值c8h9n2o5213.051。测量值213.050[m+h]+。
化合物49:在rt下将三氟乙酸酐(1.48ml,10ml)加入到处于chcl3(10ml)中的化合物48(114mg,0.54mmol)溶液中。将混合物在惰性气氛中加热至60℃持续5小时。在这段时间之后,将反应冷却至rt,并在减压下除去溶剂。向黄色油状物中加入etoh(3ml)和h2o(3ml),并将溶液在rt下搅拌2小时。减压除去溶剂,用ch2cl2(3×30ml)萃取水相。合并有机相,经mgso4干燥并减压蒸发。使用己烷/acoet(溶剂梯度为70/30至50/50),通过硅胶柱层析法纯化残余物,得到化合物49(74mg,65%)。rf(ch2cl2/meoh,95/5)=0.67;1hnmr(400mhz,cdcl3,δ):8.68(d,1h,4j2.1,h3),8.37(d,1h,4j2.1,h5),5.06(s,2h,ch2oh),4.06(s,3h,ch3co);13cnmr(100mhz,cdcl3,δ):164.3(coome),163.6(c6),155.3(c4),149.7(c2),116.4(c5),116.3(c3),64.5(ch2oh),29.5(co2ch3)。
化合物50a:在惰性气氛和rt下,将nah(17mg,0.708mmol)和硫代乙醇酸乙酯(35μl,0.320mmol)加入到处于无水dmf(1ml)中的化合物49(21.6mg,0.102mmol)的溶液中。将混合物在rt下在惰性气氛中搅拌2小时。然后在减压下除去溶剂,并向黄色油状物中加入meoh(5ml)和h2so4(200μl)。将溶液在氩气下加热至65℃持续72小时。减压除去溶剂,向残余物中加入h2o(10ml),用acoet(3×20ml)萃取水性溶液。合并有机相并经mgso4干燥,过滤并减压浓缩。使用作为洗脱剂的ch2cl2-meoh(98/2),通过硅胶柱层析法纯化残余物,得到化合物50a(8.2mg,25%)。rf(dcm/meoh,95/5)=0.35;1hnmr(400mhz,cdcl3,δ):7.88(d,1h,4j1.9,h5),7.63(d,1h,4j1.9,h3),4.69(s,2h,ch2oh),4.02(s,2h,ch2s),3.96(s,3h,ch3co),3.76(s,3h,ch3co);13cnmr(100mhz,cdcl3,δ):170.7(coome),166.4(coome),163.3(c2),152.3(c4),147.8(c6),121.2(c5),121.1(c3),65.1(ch2oh),53.3(co2ch3),48.5(co2ch3),33.6(sch2)。
化合物50b:根据与用于合成50a的过程相同的过程制备化合物50b。
化合物51a:将三乙胺(12.5μl,0.09mmol)和mscl(3.5μl,0.045mmol)加入到处于无水thf(2ml)中的化合物50a(8.2mg,0.03mmol)的溶液中。将该溶液在rt下搅拌3.5小时。在这段时间之后,在减压下除去溶剂,并将残余物溶解在ch2cl2(20ml)中。有机相用h2o(3×10ml)洗涤,经mgso4干燥,过滤并减压除去溶剂,定量得到化合物51a。rf(dcm/meoh,95/5)=0.8;1hnmr(400mhz,cdcl3,δ):7.95(d,1h,4j1.5,h5),7.52(d,1h,4j1.5,h3),5.35(s,2h,ch2oms),3.98(s,2h,ch2s),3.82(s,2h,coch3),3.77(s,2h,coch3),3.14(s,3h,sch3)。
化合物51b:根据与用于合成51a的过程相同的过程制备化合物51b。
化合物52a-52b:分别根据与用于合成14b和14c的过程相同的过程制备化合物52a-52b。
化合物53a-53b:根据与用于合成46的过程相同的过程制备化合物53a-53b。
化合物54a-54b:根据与用于合成49的过程相同的过程制备化合物54a-54b。
化合物55a-55d:根据与用于合成50a的过程相同的过程制备化合物55a-55d。
化合物56a-56d:根据与用于合成51a的过程相同的过程制备化合物56a-56d。
化合物57a:使用吡啶14a,根据与用于合成57b相同的过程制备化合物57a。
化合物57b:将无水thf(1ml)加入溴化衍生物14b(103mg,0.35mmol)中,并通过三次冻融循环使溶液脱气。向该溶液中加入乙炔衍生物(80mg,0.42mmol)和tea(0.24ml,1.75mmol)并将该溶液再次脱气。向该溶液中加入pd(dppf)cl2(30mg,0.035mmol)和cui(7mg,0.035mmol)。将该新溶液再次脱气三次,然后在65℃下在惰性气氛中搅拌。通过tlc监测反应进程。18小时后反应完成。将反应混合物冷却至rt并在减压下除去溶剂。粗产物通过硅胶柱层析法(dcm/meoh0-3%,0.1%增量)纯化,得到对应于化合物57b的黄色油状物(122mg,87%)。1hnmr(400mhz,cdcl3)δ:7.98(sl,1h),7.50(sl,1h),7.45(d,j=8.9hz,2h),6.88(d,j=8.9hz,2h),4.80(s,2h),4.65(s,2h),4.09(m,1h),3.99(sl,1h),3.86(m,1h),3.79(s,3h),1.76(d,j=14.9hz,3h),1.26(t,j=6.9hz,3h);13cnmr(100mhz,cdcl3)δ:168.9;161.0(d,j=19hz),158.8;153.3(d,j=155hz),133.8;132.9(d,j=12hz),128.1(d,j=22hz),124.2;115.1;115.0;95.7;85.6;65.2;64.3;61.3(d,j=5hz),52.5;16.5(d,j=4hz),13.5(d,j=104hz);31pnmr(162mhz,cdcl3)δ:+39.5.hrms(esi+)计算值c20h22nnao6p[m+na]+,m/z426.1082测量值:426.1063.rf=0.44(硅胶;dcm–meoh90:10)。
化合物57c:使用吡啶14c,根据与用于合成57b的过程相同的过程制备化合物57c。
化合物59a:将处于无水thf(20ml)和tea(20ml)的混合物中的乙炔衍生物(0.864g,3.1mmol)和碘衍生物14a(0.735g,2.5mmol)的溶液在搅拌下脱气20分钟。向该溶液中加入双氯双三苯基膦钯(ii)(22mg,0.031mmol)和cui(12mg,0.063mmol)。将反应在rt下搅拌12小时。通过tlc监测反应进程。在这段时间之后,反应完成。在减压下除去溶剂。向残余物中加入饱和氯化铵溶液(50ml)并用dcm(2×25ml)萃取混合物。合并有机相,用饱和氯化铵溶液(50ml)洗涤,然后用饱和nacl溶液(2×50ml)洗涤,然后用mgso4干燥。过滤后,减压除去溶剂,残余物通过硅胶柱层析法(dcm/meoh98/2)纯化,得到化合物59a,为白色固体(0.91g,82%)。ptf:143-144℃.1hnmr(500mhz,cdcl3)δ:8.04(s,1h),7.58(s,1h),7.45(d,j=8.7hz,2h),6.86(d,j=8.7hz,2h),4.83(d,j=5.4hz,2h),4.75(s,1h),4.01(t,j=6.1hz,2h),3.97(s,3h),3.31(td,j=6.1;6.1hz,2h),1.96(m,j=6.1hz,2h),1.41(s,9h)。13cnmr(125mhz,cdcl3)δ:165.4;160.8;160.1;156.2;147.3;134.1;133.8;125.8;125.4;114.9;113.9;95.9;85.4;66.1;64.8;53.2;38.1;29.7;28.6。hrms(esi+)计算值c24h28n2o6[m+h]+,m/z441.2020,测量值:441.2021.rf=0.32(硅胶,dcm-meoh96:4)。
化合物59b:将无水thf(10ml)加入溴化衍生物14b(200mg,0.68mmol)中,并通过三次冻融循环使溶液脱气。向该溶液中加入乙炔衍生物(260mg,0.75mmol)和tea(5ml),并将该溶液再次脱气。向该溶液中加入pd(pph3)4(79mg,0.068mmol)和cui(13mg,0.068mmol)。将该新溶液再次脱气三次,然后在65℃下在惰性气氛中搅拌。通过tlc监测反应的进程。1小时后反应完成。将反应混合物冷却至rt并在减压下除去溶剂。将残余物在dcm(25ml)中稀释,并用氯化铵的饱和水溶液(25ml)洗涤,然后用水(25ml)洗涤。有机相用mgso4干燥,过滤并减压浓缩,产生残余物,通过硅胶柱层析法(dcm/meoh0-5%,1%增量)纯化,得到对应于化合物59b的黄色油状物(220mg,66%)。hrms(esi+)计算值c25h34n2o6p[m+h]+,m/z489.2149测量值:489.2152.rf=0.35(硅胶,dcm–meoh,95:5)。
化合物59c:将无水thf(10ml)加入到溴化衍生物14c(142mg,0.4mmol)中,并通过三次冻融循环使溶液脱气。向该溶液中加入乙炔衍生物(147mg,0.4mmol)和tea(5ml),并将该溶液再次脱气。向该溶液中加入pd(pph3)4(46mg,0.04mmol)和cui(7.6mg,0.04mmol)。将该新溶液再次脱气三次,然后在65℃下在惰性气氛中搅拌。通过tlc监测反应进程。1小时后反应完成。将反应混合物冷却至rt并在减压下除去溶剂。将残余物在dcm(25ml)中稀释,并用氯化铵的饱和水溶液(25ml)洗涤,然后用水(25ml)洗涤。有机相用mgso4干燥,过滤并减压浓缩,产生残余物,通过硅胶柱层析法(dcm/meoh0-3%,0.5%增量)纯化,得到对应于化合物59c的黄色油状物(154mg,70%)。1hnmr(400mhz,cdcl3)δ:8.01(dd,j=6.4;2.0hz,1h),7.89(dd,j=8.4;12.4hz,2h),7.48(t,j=8.4hz,1h),7.40(d,j=8.8hz,2h),7.39(td,j=8.4;4.2hz,2h),7.33(d,j=2.0hz,1h),6.81(d,j=8.8hz,2h),4.73(s,1h),4.69(s,2h),4.08(qd,j=5.6;4.8hz,2h),3.97(t,j=6hz,2h),3.26(m,2h),1.92(q,j=6hz,2h),1.37(s,9h),1.31(t,j=5.6hz,3h);13cnmr(100mhz,cdcl3)δ:160.3(d,j=18hz),159.8;156.0;153.2(d,j=164hz);133.7;133.0(d,j=11hz);132.6(d,j=5hz);132.3(d,j=10hz);129.6(d,j=138hz);128.6(d,j=18hz);128.5(d,j=9hz);123.8(d,j=3hz);114.7;113.8;96.0;85.3(d,j=2hz);79.3;65.9;63.8;61.9(d,j=6hz);37.9;29.5;28.4;16.6;31pnmr(162mhz,cdcl3)δ:+25.6.hrms(esi+)计算值c30h36n2o6p[m+h]+,m/z551.2306测量值:551.2305.rf=0.24(硅胶,dcm–meoh,95:5)。
化合物60a:将tea(0.2ml,148μmol)在惰性气氛下滴加到在无水thf(7ml)中的醇59a(195mg,0.44mmol)溶液中。向冷却至4℃的该混合物中滴加mscl(67μl,0.84mmol)。通过tlc监测反应进程。5分钟后,反应完成。在减压下除去溶剂。将残余物溶于dcm(10ml)中,并用水(2×10ml)洗涤该溶液。有机相用mgso4干燥,过滤并减压浓缩,得到黄绿色油状物(240mg,定量)。产物60a足够纯,无需进一步纯化即在随后的合成中加以使用。lrms(esi+)计算值c25h31n2o8s[m+h]+,m/z519.1801,测量值:519.13.rf=0.6(硅胶,dcm-meoh96:4)。
化合物60b-60c:根据与用于合成60a的过程相同的过程制备化合物60b-60c。
化合物61a-61c:根据与用于合成61d的过程相同的过程制备化合物61a-61c。
化合物61d:向处于无水thf(10ml)中的化合物1(81mg,0.353mmol)溶液中,一次性加入处于无水mecn(10ml)中的化合物17a(234mg,0.706mmol)溶液和碳酸钾(195mg,1.413mmol)。将反应在60℃下搅拌过夜。通过uplc-ms(梯度a)监测反应进程。在这段时间之后,反应完成。减压浓缩反应混合物,在dcm(50ml)中稀释,用水(25ml×2)洗涤,然后用nacl(25ml)饱和水洗涤。有机相用mgso4干燥,过滤并减压浓缩。使用dcm/meoh(溶剂梯度为96/4至90/10),通过硅胶柱层析法纯化粗产物,得到白色粉末形式的化合物61d(67mg,27%)。
hplc梯度a-rt=2.91min-[m+h]+,m/z700.54–hrms(esi+)计算值c35h50n5o10+[m+h]+,m/z700.3552,测量值:700.3560-1hnmr(400mhz,cdcl3)δ:7.85(s,2h),7.69(s,1h),7.57(s,1h),4.00(s,3h,py-coome),3.97(s,10h),3(s,3h,oms),3.66(s,6h),3(m,20h),1.45(s,9h);13cnmr(100mhz,cdcl3)δ:172.48;172.39;165.90;161.48;155.68;151.13;147.55;147.43;126.27;123.70;79.29;63.56;63.40;57.15;54.84;54.66;54.35;52.88;51.84;51.80;50.09;49.50;34.07;33.93;30.11;30.06;29.68;28.56。
化合物61e-61s:根据与用于合成61d的过程相同的过程制备化合物61e-61s。
化合物62a-62c:根据与用于合成62d的过程相同的过程制备化合物62a-62c。
化合物62d:在25ml烧瓶中,将化合物61d(67mg,0.096mmol)溶解在dcm(500μl)中,得到无色溶液。将tfa(500μl,6.53mmol)一次性加入到反应混合物中。将反应在rt下搅拌30分钟。通过uplc-ms(梯度a)监测反应进程。在这段时间之后,反应完成。减压浓缩反应混合物以产生白色粉末(57mg),其用于随后的过程中。在100ml烧瓶中,将化合物60a(76mg,0.147mmol)和先前从两个组合批次(87.4mg,0.122mmol)得到的白色粉末溶解在无水mecn(30ml)中以得到无色溶液。向反应混合物中一次性加入碳酸钾(16.92mg,0.122mmol)。将反应在65℃下搅拌过夜。通过uplc-ms(梯度a)监测反应进程,在这段时间之后,反应完成。减压浓缩反应混合物,在dcm(50ml)中稀释,用水(2×40ml)洗涤。有机相用mgso4干燥,过滤并减压浓缩,得到白色粉末形式的化合物62d(10.8mg,10.6μmol,9%)。hplc梯度a-rt=2.91min-[m+h]+,m/z1022.65–hrms(esi+)计算值c54h67n7o13ag+[m+ag]+,m/z1128.3842,测量值:1128.3843。
化合物62e-62s:根据与用于合成62d的过程相同的过程制备化合物62e-62s。
化合物64a-64c:根据与用于合成64d的过程相同的过程制备化合物64a-64c。
化合物64d:在50ml烧瓶中,将化合物62d(10.8mg,10.57μmol)溶解在水(4ml)中,得到无色悬浮液。将lioh(1.291mg,0.053mmol)一次性加入到反应混合物中。将反应在rt下搅拌1小时。通过uplc-ms(梯度a)监测反应进程。在这段时间之后,脱保护完成。用1mhcl将反应混合物的ph调节至7。向反应混合物(化合物63d)中一次性加入氯化铕六水合物(5.81mg,15.85μmol)。将反应在室温下搅拌1小时,这段时间后反应完成。反应混合物通过制备型hplc(梯度b)直接纯化,得到白色粉末形式的化合物64d(6.32mg,5.74μmol,54%)。hplc梯度a-rt=2.62min-[m-2h]+,m/z1102.62–hrms(esi+)计算值c49h55euno13+[m-2h]+,m/z1100.3051,测量值:1100.3064。
化合物64e-64s:根据与用于合成64d的过程相同的过程制备化合物64e-64s。
络合物65d-e2:在25ml烧瓶中,将化合物64d(6.32mg,5.74μmol)溶解在无水dmso(1ml)中,得到无色溶液。向反应混合物中一次性加入3-氨基-1-丙磺酸(3.29mg,22.96μmol)、dipea(4μl,23μmol)和hatu(6.75mg,17.2μmol)。将反应在rt下搅拌15分钟。通过uplc-ms(梯度c)监测反应进程,在这段时间之后,反应完成。反应混合物通过制备型hplc(梯度d)直接纯化,得到白色粉末形式的可溶性络合物65d-e2(5.87mg,4.37μmol,76%)。hplc梯度c-rt=2.19min-[m-2h]+,m/z1345.26–hrms(esi+)计算值c55h70eun9o17s22+[m-h]2+,m/z672.6768,测量值:672.6769。
络合物66d-e2:在25ml烧瓶中,将络合物65d-e2(5.64mg,4.2μmol)溶解在tfa(200μl)中,得到黄色溶液。将反应在rt下搅拌30分钟。通过uplc-ms(梯度c)监测反应进程。在这段时间之后,脱保护完成。减压浓缩反应混合物并通过制备型hplc(梯度e)纯化,得到白色粉末形式的络合物66d-e2(2.16μmol,51%)。hplc梯度c-rt=1.36min-[m-2h]+,m/z1243.45–hrms(esi+)计算值c50h62eun9o15s22+[m-h]2+,m/z622.6506,测量值:622.6503。
络合物66d-e2的uv光谱、色谱和质谱分别如图1至图3所示。
我们测定了络合物66d-e2和如申请wo2014/111661中所述的合成的三天线络合物(tacn-phos-三天线和tacn-carbo-三天线)的光物理性质,所述三天线络合物的结构如下所示。
从下表可以看出,络合物66d-e2的亮度低于具有三个天线的tacn-pos-三天线和tacn-carbo-三天线的亮度。
66d-e2和tacn-phos-三天线的效率也根据以下方案进行了测试。使用本领域技术人员公知的常规技术,将每个络合物官能化为nhs酯(n-羟基琥珀酰亚胺)。使用66d-e2-nhs络合物或tacn-phos-三天线-nhs络合物标记一批抗谷胱甘肽s-转移酶(抗gst)抗体。在tacn-phos-三天线络合物(抗gst-tacn-phos-三天线)和络合物66d-e2(抗gst-66d-e2)中,抗体结合络合物的平均数目都是7。
将用铕络合物66d-e2(抗gst-66d-e2)或铕tacn-phos-三天线络合物(抗gst-tacn-phos-三天线)标记的抗gst抗体用于基于时间分辨荧光的免疫测定(如图4所示),以检测谷胱甘肽s-转移酶-生物素(gst-生物素)蛋白。在384孔板中,在一式三份的确定浓度的抗gst-66d-e2或抗gst-tacn-phos-三天线和5nm的链霉亲和素-d2(cisbiobioassaysprodno.610sadlb)的存在下,对稀释于包含bsa的磷酸盐缓冲液中的gst-生物素的浓度范围进行测量,并在孵育2小时后,在pherastarfs荧光读取器(bmg-labtech)上以htrf模式读取。结果示于图5中。用两种标记的抗体对gst-生物素进行检测。令人惊奇且预料不到的是,使用抗gst-66d-e2抗体的免疫测定的性能优于抗gst-tacn-phos-三天线,即,单天线荧光探针具有比包含三个天线的探针更高的fret能力,尽管亮度低三倍。
为了证实该结果,使用识别环磷酸腺苷(camp)的抗体进行另一免疫测定。为此目的,用66d-e2-nhs络合物标记抗camp抗体,得到平均携带6.5个络合物/抗体的抗camp-66d-e2缀合物。用tacn-carbo-三天线-nhs络合物标记同一批抗体,得到携带平均6.6个络合物/抗体的抗camp-tacn-carbo-三天线缀合物。
测试的原理描述于图6中。在使用时间分辨荧光作为检测技术的竞争性测试中使用两种缀合物。通过给出时间分辨的荧光信号,用铕络合物标记的抗camp识别与荧光团d2缀合的camp(camp,gs-dynamic试剂盒,cisbiobioassays(参考621m4pec))。camp浓度范围允许camp-d2被置换,引起信号的逐渐降低。
在牛血清白蛋白(bsa)存在下的磷酸盐缓冲液中,在camp-d2的存在下,通过在384孔板中一式三份地制备camp浓度范围,以明确的浓度使用两种抗camp-66d-e2和抗camp-tacn-carbo-三天线缀合物,并在孵育1小时后在pherastarfs盘式分析仪(bmg-labtech)上以htrf模式读取。fret信号抑制响应示于图7中。在该第二个实例中,同样令人惊奇的是,抗camp-66d-e2缀合物的性能优于抗camp-tacn-carbo-三天线缀合物的性能,即,单天线荧光探针具有比包含三个天线的探针更高的fret能力,尽管亮度减半。
由带有一个或两个苯乙炔基吡啶发色团的tacn大环组成的络合剂与镧系元素形成稳定的络合物,并可用于制备感兴趣分子的荧光缀合物。本发明所述的镧系元素络合物具有优异的光物理性质,特别是在它们的量子产率、它们的发光寿命和它们的激发光谱方面,其非常适合于在约337nm下的激光激发。此外,它们的发射光谱的谱带分布集中在620nm左右,因此,当将fret与青色素或别藻蓝蛋白型的受体(例如,由cisbiobioassays出售的xl665)一起使用时,给予络合物杰出且非常有利的性质。由于这些络合物在含有大多数二价阳离子(ca2+、mg2+等)或edta的生物介质中的高稳定性,它们的发光保持优异。
- 还没有人留言评论。精彩留言会获得点赞!