用于基于核酸实时确定疾病状态的方法和装置与流程
本发明要求于2017年7月19日提交的欧洲专利申请no.17182104.4的优先权权益,其内容通过引用整体并入本文。
本发明涉及用于在对象中确定疾病状态(例如感染)以及鉴定疾病状态的病原体(causativeagent)的方法和装置,该方法基于随时间推移确定对象中未映射至对象的核酸相对于映射至对象的核酸的量。
背景技术:
目前,用于诊断感染性疾病的方法可分为两个大的领域。一个领域涉及诊断与宿主(可能被感染)生物体有关的感染。在这一领域中,诊断本身以针对宿主是否患有感染的问题的“是”或“否”的答案形式表示。“是”表示存在感染,或者“否”表示不存在感染。诊断与感染相关的疾病的另一种方式是诊断引起感染的微生物。在这种情况下,诊断程序也仅产生“是”/“否”的答案;“是”表示患者x患有微生物y,或者“否”表示患者未患有。
如今,关注鉴定疾病病原微生物的诊断基于血液培养或pcr技术。除了纯定性的结果(“是”/“否”的答案)之外,这些诊断性方法仅能够检测限定的微生物组。对于血液培养,这是由于并非所有微生物都能在血液培养瓶内生长的事实(例如,病毒或真菌)。在基于pcr的诊断的情况下,例如由于复杂性原因,必须对限制了针对过于庞大的靶标组的特异性的引物对组进行限定。这些诊断测试不能够对可能的微生物的所有类型(例如细菌、真菌、病毒和寄生物)进行无偏的、高特异性、高灵敏度的测试。此外,尽管基于pcr的方法比血液培养更快,但是血液培养仍然是感染性疾病的第一线诊断测试。
此外,这两种方法均不能够区分共生微生物、污染物和患者所患有的真正感染原。这最终导致了许多假阳性结果。
传统的血液培养测试花费2至7天。在这段时间期间,在知晓病原微生物之前,如由主治医师根据最新的治疗指南所判断的,使用广谱抗生素对患者进行治疗。因为这一点,鉴于诊断程序较差,由于广谱抗生素的任意滥用,微生物可变得具有多种抗性。因此,为了使用合适的抗感染剂提供患者的快速且有效的治疗,有必要尽可能快速地鉴定感染原,并且至关重要的是在诊断程序期间能够区分感染原与共生微生物/污染物。
在文献中有对从患者获得的样品进行测序以鉴定其中所含微生物的实例,所述文献例如hasman等,2014,journalofclinicalmicrobiology52:139-146,其描述了对尿样品进行全基因组测序以鉴定其中所含的微生物,将该序列结果与用常规培养和鉴定获得的结果进行比较。其他文献包括:grumaz等,2016,genomemedicine8:73,其公开了对从脓毒性患者获得的样品进行下一代测序;andersson等,2013,clinmicrobiolinfect19:e405-e408,其描述了来源于阴道拭子诊断试样的dna的超深度测序;以及tumbaugh等,2009,nature457:480-484,其描述了对总的粪便dna进行鸟枪法测序(shotgunsequencing),以鉴定通常在肥胖或瘦的肠微生物组中富集的基因。这些方法简单地对非宿主核酸进行测序并将其与数据库进行比较以鉴定样品中的任何微生物。
但是,在本领域中仍然需要更有效地处理序列数据使得提供更准确的结果和/或允许更快地鉴定引起疾病的微生物,以使得可更早地开始有效的治疗。
发明概述
本发明至少部分地基于发明人这样的发现:鉴于从对象获得的生物样品中存在的核酸(但其在健康对象中通常不存在)的量,可确定对象患有疾病状态的可能性。例如,通过确定从对象获得的生物样品中映射至微生物的核酸的量,可确定对象患有由微生物引起的疾病状态(例如感染)的可能性。而且,该发现允许确定对象患有癌症的可能性,并且在监测癌症治疗中特别有用。在一个实施方案中,这种可能性通过以下来确定:基于映射至(分配给)特定微生物的序列读取的总数和可映射至(分配给)物种的所有序列读取的总数,计算在对象中发现映射至特定微生物的核酸序列的概率的显著性评分(significancescore),所述可映射至(分配给)物种的所有序列读取的总数包括映射至与对象相同的物种的读取的数目和映射至样品中任何微生物的读取的数目。基本上基于映射至特定微生物的序列读取的数目与映射至从对象获得的生物样品中存在的物种的序列读取的总数之比,这种显著性评分可随时间推移进行计算,即随着映射的读取总数的增加(随着越来越多的序列读取被获得并映射至物种)实时地计算。
在一个实施方案中,本发明涉及用于在对象中确定微生物的存在的方法,在一个实施方案中,所述方法包括确定映射至特定微生物的基因组的序列读取的数目和映射至物种(包括与对象相同的物种)的基因组的序列读取的数目。从对获得自对象的生物样品中存在的核酸进行测序而获得的序列读取可与一个或更多个数据库进行比较,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息。因此,可确定映射至物种(包括宿主物种和任何微生物)的序列读取的数目和映射至特定微生物的序列读取的数目。在一个实施方案中,所述方法还包括计算针对特定微生物的显著性评分,该显著性评分基于映射至该特定微生物的序列读取的数目和映射至物种的读取的总数。由于确定步骤可随时间推移而进行,因此,随着获取和映射序列读取,也可随时间推移执行该显著性评分计算。同样,在已经获得序列读取但尚未进行比较并且尚未将其映射至物种的一些实施方案中,在序列读取与一个或更多个数据库中的遗传信息进行比较时,也可随时间推移执行该计算。
本发明涉及用于在对象中确定微生物的存在的方法,其包括以下步骤:(a)对从对象获得的生物样品中存在的核酸进行测序以获得多个核酸序列读取;(b)将步骤(a)中获得的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息;以及(c)随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目。
本发明还涉及用于在对象中确定微生物的存在的方法,其包括:(a)(a)将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息,其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而获得的;以及(b)随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目。
本发明还涉及用于在对象中确定微生物的存在的方法,其包括随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目的步骤,其中所比较的序列读取是通过以下获得的:将所产生的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息,并且其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而产生的。
在本发明的一个实施方案中,所述方法还包括基于映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目,计算在对象中发现映射至特定微生物的所比较的序列读取的概率的显著性评分。
本发明还涉及用于在对象中确定微生物的存在的方法,其包括以下步骤:基于映射至特定微生物的序列读取的数目和映射至物种的序列读取的数目,随时间推移计算在对象中发现映射至特定微生物的序列读取的概率的显著性评分,其中映射至特定微生物的序列读取和映射至物种的序列读取是通过以下获得的:将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息,并且其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而产生的。
本发明还涉及用于在对象中确定微生物的存在的方法,其包括:(a)随时间推移确定映射至特定微生物的序列读取的数目和映射至物种的序列读取的数目的步骤,其中序列读取是通过以下获得的:将序列读取与一个或更多个数据库进行比较以确定序列读取是否映射至一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息,并且其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而产生的;以及(b)基于映射至特定微生物的序列读取的数目和映射至物种的序列读取的数目,计算在对象中发现映射至特定微生物的序列读取的概率的显著性评分。
在本发明的多个实施方案中,可执行这样的方法,其中紧接着(即基本上同时)对核酸进行测序之后比较序列读取,以将读取映射至物种并计算显著性评分,或者可在早于比较/确定/计算步骤的任何时间时进行测序,以使得测序结果被存储,然后存储的测序结果可用于将测序的读取与一个或更多个数据库进行比较,并且例如,允许计算显著性评分。
在一个实施方案中,随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目的步骤意指对可映射至特定微生物的所比较的读取的数目进行计数,并对可映射至物种的所比较的读取的数目进行计数,所述可映射至物种的所比较的读取即不仅是映射至特定微生物的读取,而且还是映射至对象、以及映射至样品中存在的任何其他微生物的读取。可能由于降解、长度太短或来自不存在于一个或更多个数据库中的微生物而不能够映射至物种的那些序列读取未用于本发明。优选地,并非所有的序列读取均用于本发明,仅能够映射至物种的那些序列读取用于本发明。
在一个实施方案中,当针对特定微生物的显著性评分达到或超过阈值时,确定特定微生物存在于对象中,或者当针对特定微生物的显著性评分达到或超过阈值时,确定特定微生物与在对象中引起疾病相关。在另一些实施方案中,显著性评分超过阈值越多,对象中微生物的负荷越高,这可反映出更严重的感染状态。在一个实施方案中,设定阈值以使关于特定微生物在对象中引起疾病的相关性的假阳性和假阴性数目最小化。
在另一个实施方案中,当针对特定微生物的显著性评分在少数序列读取映射至物种就超过阈值时,由于微生物的存在而引起的疾病可被认为是严重的。在该实施方案的背景下,“少数(few)”是指这样的事实,即已对通过对样品中的核酸进行测序而产生的测序的读取的并非全部(即一部分)进行了比较和映射,但是已经达到或超过阈值。所比较和映射的读取的部分可以是所有所比较和映射的读取的1%、2%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%或95%。优选地,“少数”是指少于所有所比较和映射的读取的50%、45%、40%、35%、30%、25%、20%、15%、10%、5%、2%、1%。“少数”也可以是固定数目的读取,例如少于100、1,000、10,000或100,000个读取。
在一个实施方案中,可随时间推移执行本发明的方法直至如下的时间点:所提供的信息,例如,映射至对象/特定微生物的读取的数目或另外的参数(包括下文所述的参数),允许在一定程度的确定性下确定对象患有或未患有疾病状态或者感染有或未感染有一种或更多种微生物,以及允许鉴定一种或更多种微生物或者癌症类型。一旦达到这一点,就可停止所述方法,因为不需要提供另外的信息来在对象中确定微生物或疾病状态的存在。
随时间推移映射至特定微生物的读取的数目和映射至物种的读取的数目可用于随时间推移产生参数,该参数不仅可用于确定例如特定微生物是否与对象中的疾病状态相关,而且也可用于允许比较两个或更多个患者之间的(相同原因的)疾病状态。换句话说,在两个患者之间映射至物种的读取的数目相同但映射至特定微生物的读取的数目不同(或多或少)的情况下,这种不同可表明两个患者之间特定微生物的负荷/量的不同。例如,如果一个对象在106个映射至物种的读取中有1个特定微生物的读取,而第二个对象在5×105个映射至物种的读取中有1个同一特定微生物的读取,那么可得出结论:所述微生物不仅存在于第二个对象中,而且第二个对象具有更高的感染负荷/水平。
此外,该参数可在所述方法期间在任何时间时实时(随时间推移)产生,而不只是在已经比较了所有序列读取和已经映射了所有所比较的读取的终点处产生。因此,如果在已经比较和映射了全部读取的仅一部分的时间点处,观察到相对于与在对照样品中观察到的相同数目的映射至物种的读取,一个对象具有5倍数目的映射至特定微生物的读取,则可在比较和映射所有测序的读取之前的较早时间点处停止所述方法,因为很显然,具有5倍更多读取的患者很可能患有由于特定微生物而引起的疾病状态(感染)。
在测序、比较和映射步骤期间随时间推移产生该参数以使得可在分析结束(即在其中,样品中的所有核酸均已进行测序并且所有读取均已进行比较和映射)之前停止所述方法的能力,相比于不能这样停止的方法,有利地允许节省时间和资源。例如,通常样品中所有核酸的测序、比较和映射步骤可花费多至30小时或更多。然而,本发明允许显著地减少该时间,例如,在一些情况下减少10小时或更多,以使得可节省10小时的测序和/或计算机时间。此外,由于可更快地诊断对象,因此可更快地开始合适的治疗,从而导致对象存活的更高的可能性。这也允许不浪费没有适当地靶向以治疗感染或疾病状态的药物,例如,给予针对病毒感染的抗生素或给予微生物对其具有抗性的抗生素。
本发明还涉及用于在对象中确定疾病状态的存在的方法,其包括:(a)对从对象获得的生物样品中存在的核酸进行测序以获得多个核酸序列读取;(b)将步骤(a)中获得的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至对照对象,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息;以及(c)随时间推移确定映射至和未映射至对照对象的所比较的序列读取的数目。本发明还涉及用于在对象中确定疾病状态的存在的方法,其包括:(a)将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至对照对象,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息,其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而获得的;以及(b)随时间推移确定映射至和未映射至对照对象的所比较的序列读取的数目。本发明还涉及用于在对象中确定疾病状态的存在的方法,其包括随时间推移确定映射至和未映射至对照对象的所比较的序列读取的数目的步骤,其中所比较的序列读取是通过以下获得的:将所产生的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至对照对象,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息,并且其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而产生的。
在一个实施方案中,所述方法还包括基于未映射至对照对象的所比较的序列读取的数目和能够进行映射(例如映射至对照对象)的所比较的序列读取的数目,计算在对象中发现未映射至对照对象的所比较的序列读取的概率的显著性评分。
本发明还涉及用于在对象中确定疾病状态的存在的方法,其包括以下步骤:基于未映射至对照对象的序列读取的数目和映射至对照对象的序列读取的数目,随时间推移计算在对象中发现未映射至对照对象的序列读取的概率的显著性评分,其中映射至对照对象的序列读取和未映射至对照对象的序列读取是通过以下获得的:将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至对照对象,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息,并且其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而产生的。
在一个实施方案中,当显著性评分达到或超过阈值时,确定疾病状态存在于对象中。本文中使用的术语“未映射至对照对象的所比较的序列读取”并不总是意指该序列与对照对象的序列不高度相似或实际上不相同,尽管其通常可如此。例如,在疾病状态是由对象的核酸序列中的点突变引起的疾病状态的一个实施方案中,具有这样的点突变的序列读取被认为未映射至对照对象,即使该读取的所有其他核苷酸与对照对象相同。此外,在一个实施方案中,当比较序列读取时,可参照已知的基因组多态性,例如参照单核苷酸多态性,以使得这些差异不被认为是对象的测序的读取中的突变。
在本发明的一个实施方案中,疾病状态是癌症,优选是由遗传异常(例如点突变、缺失、插入或插失(indel))引起的癌症。在另一个实施方案中,疾病状态是由微生物引起的感染,优选地其中所述微生物是病毒、细菌、真菌或寄生物。
在疾病状态是癌症的一个实施方案中,本发明的方法还可用于监测癌症的治疗以及监测在一轮治疗之后癌症的再现。例如,已经被诊断为患有癌症的对象进行治疗,例如手术切除肿瘤。可建立肿瘤遗传信息的数据库,并且可对从对象获得的核酸进行测序,并且可将读取与一个或更多个数据库进行比较,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和肿瘤的遗传信息。然后将所比较的读取映射至对照对象或肿瘤数据库,以使得基于映射至癌症基因组的读取的数目和映射至癌症基因组和对照基因组的读取的数目,计算根据本发明的显著性评分,从而允许确定癌症的存在,即癌症的再现。类似地,可在治疗期间获得样品,并且可计算评分以确定治疗是否有效。
在疾病状态是由微生物引起的感染的一个实施方案中,本发明的方法也可用于监测感染的治疗和/或监测感染的再现。在这样的实施方案中,生物样品在治疗期间和/或在治疗之后从对象获得,并且遵循如上所述的方法,以使得基于映射至微生物的读取的数目和映射至物种的读取的数目,计算显著性评分。
在某些实施方案中,生物样品可选自全血、血清、血浆、羊水、滑液、液体(liquor)、组织或细胞涂片、组织或细胞拭子、尿、组织、痰、粪便、胃肠道分泌物、淋巴液和灌洗液(lavage)。
在某些实施方案中,对象是脊椎动物,优选哺乳动物,例如人、狗、猫、猪、马、牛、绵羊、山羊、小鼠或大鼠,优选地对象是人。
在一个实施方案中,使用超深测序法或高通量测序法进行测序。在本发明的一些优选实施方案中,测序通过分子高通量序列分析进行,即通过下一代测序或第三代测序进行,例如通过illumina/solexa或oxfordnanopore方法进行。
在本发明的一个实施方案中,当确定特定微生物或疾病状态存在于对象中时,所述方法还包括向对象施用已知治疗由特定微生物引起的疾病或疾病状态的药物活性化合物。此外,一旦鉴定了引起感染性疾病的微生物,就可确定其是否对任何类型的抗生素/抗感染剂具有抗性,从而使治疗有效。在一个实施方案中,可在确定微生物是否对任何类型的抗生素/抗感染剂具有抗性之前耗尽样品中对象的核酸。
在一个具体的实施方案中,用于在对象中诊断由微生物引起的感染性疾病的方法包括基于映射至特定微生物的序列读取的数目和映射至物种的所比较的序列读取的数目,随时间推移计算在对象中发现映射至特定微生物的序列读取的概率的显著性评分,其中当针对特定微生物的评分达到或超过阈值时,确定特定微生物引起感染性疾病,并且其中映射至特定微生物的序列读取和映射至物种的序列读取是通过以下获得的:将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息,并且其中序列读取是通过对从对象获得的生物样品中存在的核酸进行测序而产生的。
在一个具体的实施方案中,用于在对象中诊断由微生物引起的感染性疾病的方法包括:(a)对从对象获得的生物样品中存在的核酸进行测序以获得多个核酸序列读取;(b)将步骤(a)中获得的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息;(c)随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目;以及(d)基于映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目,计算在对象中发现映射至特定微生物的所比较的序列读取的概率的显著性评分,其中当针对特定微生物的评分达到或超过阈值时,确定特定微生物引起感染性疾病。
本发明涉及在对象中治疗由微生物引起的感染或疾病的方法,其包括:(a)根据任意前述用于在对象中确定微生物的存在的方法,在对象中确定针对特定微生物的显著性评分;以及(b)当针对特定微生物的显著性达到或超过阈值时,向对象施用抑制特定微生物的生长的化合物。本发明还涉及在对象中治疗由微生物引起的感染或疾病的方法,其包括向对象施用抑制显著性评分达到或超过阈值的微生物的生长的化合物,其中显著性评分根据本文中所述的任意前述用于在对象中确定微生物的存在的方法来计算。
本发明还包括存储程序代码的计算机可读存储介质,其包含指令,所述指令当由处理器执行时实施本发明的方法;以及含有被配置为实施本发明方法的处理器的计算机系统,例如现场可编程门阵列。
发明详述
尽管下面详细描述了本发明,但是应理解,本发明不限于本文中所述的特定方法、方案和试剂,因为这些可变化。还应理解,本文中使用的术语仅用于描述特定实施方案的目的,并不旨在限制本发明的范围,本发明的范围仅由所附权利要求书限制。除非另有限定,否则本文中使用的所有技术和科学术语具有本领域普通技术人员通常理解的相同含义。
在下文中,将描述本发明的要素。这些要素伴随具体实施方案列举,但是,应理解,它们可以以任何方式及任何数目组合以产生另外的实施方案。多种所描述的实例和优选实施方案不应被解释为将本发明限于仅是明确描述的实施方案。该描述应理解为支持和涵盖将明确描述的实施方案与任何数目的所公开的和/或优选的要素组合的实施方案。此外,除非上下文中另有指明,否则应认为本申请中所有描述的要素的任何排列和组合均由本申请的说明书所公开。
优选地,本文中使用的术语如“amultilingualglossaryofbiotechnologicalterms:(iupacrecommendations)”,h.g.w.leuenberger,b.nagel和h.
除非另有指明,否则本发明的实践将采用在本领域文献中说明的生物化学、细胞生物学、免疫学和重组dna技术的常规方法(参见例如molecularcloning:alaboratorymanual,2ndedition,j.sambrook等eds.,coldspringharborlaboratorypress,coldspringharbor1989)。
在整个说明书和随后的权利要求书中,除非上下文中另有要求,否则词语“包括/包含”及其变化形式将被理解为意指包括所陈述的成员、整数或步骤或者成员、整数或步骤的组,但不排除任何其他成员、整数或步骤或者成员、整数或步骤的组,尽管在一些实施方案中,这样的其他成员、整数或步骤或者成员、整数或步骤的组可被排除,即,主题在于包括所陈述的成员、整数或步骤或者成员、整数或步骤的组。除非本文中另有指明或与上下文明显矛盾,否则在描述本发明的上下文中(尤其在权利要求书的上下文中)使用的没有数量词修饰的名词表示一个/种或更多个/种。本文中值的范围的记载仅旨在作为单独地指落入该范围内的各独立值的速记方法。除非本文中另外指明,否则各单独的值均并入说明书中,如同其在本文中单独记载一样。
除非本文中另有指明或另外地与上下文明显矛盾,否则可以以任何合适的顺序执行本文中所描述的所有方法。本文中提供的任何和所有实例或示例性语言(例如,“例如”)的使用仅旨在更好地说明本发明,并且不对另外要求保护的本发明的范围进行限制。本说明书中的任何语言均不应被解释为表示对实施本发明而言必要的任何非要求保护的要素。
本说明书通篇中引用了数篇文献。本文中所引用的每篇文献(包括全部专利、专利申请、科学出版物、制造商的说明书、指南等),无论是在上文中的还是在下文中的,均通过引用整体并入本文。本文中的任何内容均不应被解释为承认本发明无权凭借在先发明而先于这样的公开内容。
如上所述,本发明基于与能够映射至例如物种/正常基因组的读取的总数相关的映射至例如特定微生物或癌症基因组的序列读取的数目。因此,本发明提供了用于诊断和区分共生体/污染物与最可能的感染病原体的基础。有利地,本发明提供了至少以下内容:
a)对所获得的生物样品不做任何假设的无偏方法,
b)能够分辨共生体/污染物与感染原的方法,
c)实时提供在给定的时间处样品中所鉴定的所有微生物的结果的方法,
d)在测序期间实时产生数据的方法,
e)在处理数据的同时实时提供信息的方法,
f)一旦确定微生物对疾病状态而言显著/与疾病状态相关,就可在分析整个数据集的仅一小部分之后停止的方法,
g)产生允许比较同一疾病状态的两个或更多个生物样品的参数的方法,以及
h)使临床医生和研究人员能够在被同一微生物感染的患者中比较由于微生物引起的感染的严重程度的方法。
本发明的另一个优点是能够检测由多种微生物引起的感染,以及能够确定哪种微生物是主要的病原体和哪些是伴随病原体,尽管这些病原体均可对感染/疾病的状态起重要作用。
术语“对象”、“个体”、“生物体”或“患者”可互换使用,并且涉及脊椎动物,优选哺乳动物。例如,在本发明上下文中的哺乳动物是:人;非人灵长类动物;家养动物,例如狗、猫、绵羊、牛、山羊、猪、马等;实验室动物,例如小鼠、大鼠、兔、鱼、豚鼠等;以及圈养动物,例如动物园的动物。术语“动物”还包括人。优选地,术语“对象”、“个体”、“生物体”或“患者”是指雄性和雌性哺乳动物,特别地男人和女人。对象可以是任何年龄的,包括新生儿(例如从出生至约6个月)、婴儿(例如从约6个月至约2岁)、儿童(例如从约2岁至约10岁)、青少年(例如从约10岁至约21岁)和成人(例如约21岁及更大)。
在某些实施方案中,对象可以是例如由于服用免疫抑制药物或正在经历需要抑制或破坏天然免疫系统/功能的移植而免疫受损的。另一些对象可以是患有慢性或系统性感染的对象。在一些具体的实施方案中,对象可被怀疑患有或患有脓毒症(sepsis)、心内膜炎(endocarditis)、关节(包括人工关节)感染或软组织感染。在一个实施方案中,对象是被怀疑患有或患有脓毒症的新生儿。在另一个实施方案中,怀疑的感染是在妊娠期间在子宫内,例如羊膜内感染(绒毛膜羊膜炎)。
在本发明的上下文中,“对照”或“对照组”分别是指来自对象的生物样品或来自对象的组的样品,所述对象是健康的或被认为是健康的,即未患有疾病或至少未患有与所测试对象相同的疾病。优选地,对照或对照组包含来自健康个体的样品,所述个体以多种方式与对象匹配,例如,相似的年龄、相同的性别、相同的社会阶层或相同的民族群体或生活在国家、州或城市的基本上相同的区域。
在本发明的上下文中,术语“健康的”意指没有表现出特定疾病的任何迹象的对象,并且优选地是目前未正在发生疾病的对象。例如,健康的对象没有显示出感染或疾病的迹象,但其仍然是多种共生微生物物种的宿主。优选地,对象不是被感染的对象,而是处于感染不明显的感染阶段的对象。
本文中使用的“生物样品”包括从对象,例如从对象的身体获得的任何生物样品。这样的生物样品的一些实例包括:全血;血液级分,例如血浆、血清;组织涂片或拭子;痰;支气管抽吸物;尿;精液;粪便;胆汁;胃肠道分泌物;生殖系统分泌物;羊水;滑液;淋巴液;液体;骨髓;器官抽吸物;以及组织活检,包括穿孔活检。任选地,生物样品可获自患者的黏膜。术语“生物样品”还可包括经处理的生物样品,例如级分或分离物,例如核酸或分离的细胞。优选地,生物样品包含核酸,例如基因组dna或mrna,以使得可确定核酸的序列。在一个实施方案中,生物样品可以是从表现出疾病状态的迹象(例如表现出感染的迹象)的组织获得的样品。在一个优选的实施方案中,生物样品是获自对象的血液或血浆。根据本发明的方法对样品进行分析,并且在该方法期间或之后,样品通常不返回身体。在大多数实施方案中,为了实施本发明的方法,对象身体的存在不是必需的。
在一个实施方案中,生物样品是血浆,优选直接从对象获得。血浆优选是无细胞的,优选主要/大部分是无细胞的,例如少于10,000、1,000、100或10个细胞/ml。生物样品,例如血浆,可包含游离的循环核酸,其包含对象的核酸和非对象的核酸,例如微生物的那些核酸。在一个实施方案中,可稀释或浓缩生物样品。在另一个实施方案中,在测序之前对样品进行处理,优选在测序之前对样品进行纯化以除去细胞组分,例如脂质和蛋白质。在一个实施方案中,在测序之前对生物样品进行处理,以使得仅对无细胞的核酸进行测序。
可从中获得生物样品的患者组织包括但不限于:喉、口、鼻、胃、肠、皮肤、关节、肝、胰、肺、神经元、宫颈、阴道、子宫、尿道、直肠、阴茎和肌肉。可结合本发明使用用于从患者和/或从合适的组织获得生物样品的任何合适的方法。
术语“体内”涉及对象内的情况。
术语“基因组”涉及生物体或细胞的染色体中的遗传信息的总量。
术语“外显子组(exome)”是指由外显子形成的生物体的基因组部分,其是所表达基因的编码部分。外显子组提供用于合成蛋白质及其他功能性基因产物的遗传蓝图。其是基因组中功能最相关的部分,因此,其最有可能促成生物体的表型。人基因组的外显子组估计占总基因组的1.5%(ng等,2008,plosgen4(8):1-15)。
术语“转录物组”涉及一个细胞或细胞群中产生的所有rna分子的集,所述rna分子包括mrna、rrna、trna以及其他非编码rna。在本发明的上下文中,转录物组意指给定个体的一个细胞、细胞群或所有细胞在某一时间点产生的所有rna分子的集。
术语“遗传物质”包括分离的核酸(dna或rna)、双螺旋的部分、染色体的部分、或者生物体或细胞的整个基因组,特别是其外显子组或转录物组。
根据本发明,“核酸”优选是脱氧核糖核酸(dna)或核糖核酸(rna)。核酸包括基因组dna、cdna、mrna、重组产生的分子和化学合成的分子。核酸可作为单链分子或双链分子和线性或共价环状闭合的分子以及它们的混合物存在。核酸可被分离。优选地,核酸是游离的循环dna和/或rna分子。在一个实施方案中,术语“核酸”也被理解为意指“核酸序列”。此外,在测序之前,可对核酸进行处理,例如对其进行富集或扩增。在从样品获得的核酸是rna的情况下,可将rna反转录为dna用于测序,也可对rna本身进行测序。
术语“突变”是指与参照相比核酸序列的改变或差异(核苷酸替换、添加或缺失)。除生殖细胞(精子和卵子)之外,身体的任何细胞都可发生“体细胞突变”,因此不会传递给儿童。这些改变可(但不总是)导致癌症或其他疾病。优选地,突变是非同义突变。术语“非同义突变”是指导致翻译产物中氨基酸改变(例如氨基酸替换)的突变,优选核苷酸替换。
根据本发明,术语“突变”包括点突变、插失、融合、染色体碎裂(chromothripsis)和rna编辑。
根据本发明,术语“插失”描述了一种特殊的突变种类,其定义为导致共定位的插入和缺失以及核苷酸的净增加或损失的突变。在基因组的编码区中,除非插失的长度是3的倍数,否则其产生移码突变。插失可与点突变形成对比,其中插失从序列中插入和缺失核苷酸,点突变是替代一个核苷酸的替换形式。
根据本发明,术语“染色体碎裂”是指通过单个灾难性事件(devastatingevent)基因组的特定区域被破碎并随后拼接在一起的遗传现象。
融合可产生由两个先前分离的基因形成的杂合基因。其可由于易位、中间缺失或染色体倒位而发生。通常,融合基因是癌基因。致癌性融合基因可导致具有新功能或不同于两个融合配偶体功能的基因产物。或者,原癌基因与强启动子融合,并且由此,致癌性功能被设定为通过由上游融合配偶体的强启动子引起的上调而起作用。致癌性融合转录物还可通过反式剪接或通读事件产生。
在本发明的上下文中,术语“测序”意指确定至少一种核酸的序列,并且其包括用于确定至少一种核酸的链中碱基顺序的任何方法。一种优选的测序方法是高通量测序,例如下一代测序或第三代测序。
为了说明目的,术语“下一代测序”或“ngs”在本发明的上下文中意指全部高通量测序技术,其与称为sanger化学的“常规”测序方法形成对比,通过将整个基因组断裂成小碎片来沿着整个基因组平行地随机阅读核酸模板。这样的ngs技术(也称为大规模平行测序技术)能够在非常短的时间段内,例如在1至2周内,优选地在1至7天内或者最优选地在不到24小时内递送整个基因组、外显子组、转录物组(基因组的所有转录序列)或甲基化组(基因组的所有甲基化序列)的核酸序列信息并且在原理上实现单细胞测序方法。在本发明的上下文中可使用可商购的或文献中提及的多种ngs平台,例如zhang等,2011,theimpactofnext-generationsequencingongenomics.j.genetgenomics38:95-109中;或者voelkerding等,2009,nextgenerationsequencing:frombasicresearchtodiagnostics,clinicalchemistry55:641-658中详细描述的那些。这样的ngs技术/平台的非限制性实例为:
1)在例如roche联合公司454lifesciences(branford,connecticut)的gs-flx454genomesequencertm中实施的称为焦磷酸测序的边合成边测序技术,其首先在ronaghi等,1998,asequencingmethodbasedonreal-timepyrophosphate,science281:363-365中进行了描述。这项技术使用乳液pcr,其中通过剧烈涡旋将单链dna结合珠包封在由油包围的包含pcr反应物的水性胶束中,以用于进行乳液pcr扩增。在焦磷酸测序过程期间,随着聚合酶合成dna链,记录在核苷酸并入期间从磷酸分子发射的光。
2)由solexa(现在为illuminainc.,sandiego,california的一部分)开发的边合成边测序方法,其基于可逆染料-终止剂并且例如在illumina/solexagenomeanalyzertm中和在illuminahiseq2000genomeanalyzertm中实施。在这项技术中,将全部四种核苷酸与dna聚合酶一起同时添加到流式细胞通道中的寡聚物引发的簇片段中。桥接扩增用所有四种荧光标记的核苷酸延伸簇链以用于测序。
3)在例如appliedbiosystems(现在为lifetechnologiescorporation,carlsbad,california)的solidtm平台中实施的边连接边测序方法。在这项技术中,根据测序位置标记固定长度的所有可能寡核苷酸的库。使寡核苷酸退火并连接;dna连接酶对匹配序列的优先连接产生在该位置处核苷酸的信息的信号。在测序之前,通过乳液pcr扩增dna。将各自均仅包含相同dna分子的拷贝的所得珠沉积在载玻片上。作为第二个实例,doversystems(salem,newhampshire)的polonatortmg.007平台还采用通过使用随机排列的基于珠的乳液pcr来扩增dna片段用于平行测序的边连接边测序方法。
4)例如如在pacificbiosciences(menlopark,california)的pacbiors系统或在helicosbiosciences(cambridge,massachusetts)的heliscopetm平台中实施的单分子测序技术。这项技术的独特特征是其能够不经扩增对单dna或rna分子进行测序,定义为单分子实时(single-moleculerealtime,smrt)dna测序。例如,heliscope使用高灵敏荧光检测系统来直接随着每个核苷酸合成对其进行检测。visigenbiotechnology(houston,texas)已经开发了基于荧光共振能量转移(fluorescenceresonanceenergytransfer,fret)的类似方法。其他基于荧光的单分子技术来自u.s.genomics(geneenginetm)和genovoxx(anygenetm)。
5)用于单分子测序的纳米技术,其中使用例如布置在芯片上以监测在复制期间聚合酶分子在单链上的移动的多种纳米结构。基于纳米技术的方法的非限制性实例是oxfordnanoporetechnologies(oxford,uk)的gridontm平台、由nabsys(providence,rhodeisland)开发的杂交辅助纳米孔测序(hanstm)平台、以及称为组合探针-锚连接(cpaltm)的具有dna纳米球(dnananoball,dnb)技术的基于专有连接酶的dna测序平台。
6)用于单分子测序的基于电子显微术的技术,例如由lightspeedgenomics(sunnyvale,california)和halcyonmolecular(redwoodcity,california)开发的那些。
7)基于检测在dna聚合期间释放的氢离子的离子半导体测序。例如,离子激流系统(iontorrentsystem)(sanfrancisco,california)使用微加工孔的高密度阵列以大规模并行方式进行这一生物化学过程。每个孔容纳不同的dna模板。在孔之下是离子灵敏层并且在其之下是专有离子传感器。
可用于本发明的上下文中的其他测序方法包括隧穿电流测序(tunnelingcurrentssequencing)(xu等,2007,theelectronicpropertiesofdnabases,small3:1539-1543,diventra,2013,fastdnasequencingbyelectricalmeansinchescloser,nanotechnology24:342501)。特别优选的下一代测序(next-generationsequencing,ngs)方法包括illumina、iontorrent和nanopore测序。
优选地,dna和rna制备物充当ngs的起始物质。这样的核酸可容易地从生物样品获得,例如从血液或新鲜的、快速冷冻的或福尔马林固定的组织样品或者从新鲜分离的细胞或者从患者外周血中存在的循环肿瘤细胞(circulatingtumorcell,ctc)获得。可从正常的体细胞组织提取正常的(未突变的)基因组dna或rna,但是种系细胞是优选的。种系dna或rna可从患有非血液学恶性肿瘤的患者中的外周血单个核细胞(pbmc)提取。虽然所提取的核酸可为高度片段化的,但是其仍然适合于ngs应用。
用于外显子组测序的数种靶向ngs方法在文献中进行了描述(有关综述,参见例如teer和mullikin,2010,humanmolgenet19:r145-51),其所有均可与本发明联合使用。这些方法(描述为例如基因组捕获、基因组分隔、基因组富集等)中有很多使用杂交技术并且包括基于阵列(例如,hodges等,2007,natgenet39:1522-1527)和基于液体的(例如,choi等,2009,procnatlacadsciusa106:19096-19101)杂交方法。用于dna样品制备和后续外显子组捕获的市售试剂盒也是可获得的:例如,illuminainc.(sandiego,california)提供truseqtmdna样品制备试剂盒和外显子组富集试剂盒truseqtm外显子组富集试剂盒。
一旦对核酸进行了测序,就可将所得的序列(测序的读取)与包含优选来自多个物种的遗传信息的一个或更多个数据库进行比较,以使得可确定测序的读取来自特定物种,例如对象和/或来自特定微生物,这允许确定映射至特定微生物的测序的读取的数目和映射至物种(即映射至对象以及映射至任何微生物)的测序的读取的数目。如上所说明的,不能映射至任何物种的测序的读取未用于本发明。用于映射测序的读取以提供有关其起源物种的信息的方法是本领域中公知的,并且任何这样的合适的方法均可与本发明结合使用。例如,可使用wood和salzberg,2014,genomebiol15:r46中描述的kraken超快宏基因组学序列分类方法。另一示例性方法是nextgenmap,其描述于sedlazeck等,2013,bioinformatics29:2790-2791中。另一示例性方法是如在naccache等,2014,genomeres24:1180-1192中描述的用于从临床样品的下一代测序中进行超快病原体鉴定的云兼容性生物信息学途径。本领域中已知的并且可用于本发明的另外方法包括但不限于huson等,2007,genomeres17:377-386;freitas等,2015,nuclacidsres43:e69;和kim等,2016,genomeres26:1721-1729中描述的那些。
在本发明的某些实施方案中,为了降低检测和比较序列中假阳性结果的数目,优选一式两份地确定/比较序列。因此,优选地,确定生物样品中的核酸序列两次、三次或更多次。在一个实施方案中,确定肿瘤样品的核酸序列两次、三次或更多次。还可通过至少一次地确定基因组dna中的序列和至少一次地确定所述样品的rna中的序列来确定序列多于一次。例如,通过确定样品的重复之间的变化,可估计作为统计量的预期假阳性率(fdr)突变。样品的技术重复应产生相同的结果,并且在该“相同间比较(samevs.samecomparison)”中的任何检测到的突变均为假阳性。此外,可使用机器学习方法来将多个质量相关计量(例如,覆盖率或snp质量)组合成单质量评分。对于给定的体细胞变异,可对具有超标质量评分的所有其他变异进行计数,这使得能够对数据集中的所有变异进行排序。
在本发明的上下文中,术语“数据库”可涉及有组织的数据集合,优选地如电子归档系统(electronicfilingsystem);以及涉及非结构化的数据集合,例如数据湖(datalake),其是以其自然格式存储的数据的系统或数据存储库。数据湖可以是所有企业数据的单个存储,其包含源系统数据和用于任务(例如报告、可视化、分析和机器学习)的转换数据的原始拷贝。在一些实施方案中,数据湖可包含来自关系数据库的结构化数据(行和列)、半结构化数据(csv、日志、xml、json)、非结构化数据(电子邮件、文档、pdf)和/或二进制数据(图片、音频、视频)。在一个实施方案中,序列数据库是一种类型的数据库,其由存储在计算机上的计算机化(“数字”)核酸序列、蛋白质序列或其他聚合物序列的集合构成。优选地,数据库是核酸序列的集合,即来自许多物种的遗传信息的集合。遗传信息可来源于物种的基因组和/或外显子组和/或转录物组。可用于本发明的一些示例性核酸数据库包括但不限于:国际核苷酸序列数据库(internationalnucleotidesequencedatabase,insd)、日本dna数据库(国立遗传学研究所)、embl(欧洲生物信息学研究所)、genbank(国家生物技术信息中心)、生物信息采集器(bioinformaticharvester)、基因疾病数据库(genediseasedatabase)、snpedia、微生物基因组学和宏基因组学camera资源、ecocyc(描述模式生物大肠杆菌(e.coli)k-12的基因组和生化机器的数据库)、ensembl(提供针对人、小鼠、其他脊椎动物和真核生物基因组的自动注释数据库)、ensembl基因组(通过一组统一的交互式和程序化界面(使用ensembl软件平台),提供针对细菌、原生生物、真菌、植物和无脊椎动物后生动物的基因组规模数据)、外显子组集成联合(exomeaggregationconsortium,exac)(来自广泛多种的大规模测序项目的外显子组测序数据(broadinstitute))、patric(pathosystems资源集成中心)、mgi小鼠基因组(jacksonlaboratory)、doe联合基因组研究所的jgi基因组(提供许多真核生物和微生物基因组的数据库)、国家微生物病原体数据资源(针对如下病原体的注释基因组数据的人工整理数据库:弯曲杆菌(campylobacter)、衣原体(chlamydia)、嗜衣原体(chlamydophila)、嗜血杆菌(haemophilus)、李斯特菌(listeria)、支原体(mycoplasma)、奈瑟菌(neisseria)、葡萄球菌(staphylococcus)、链球菌(streptococcus)、密螺旋体(treponema)、脲原体(ureaplasma)和弧菌(vibrio))、regulondb(细胞大肠杆菌k-12的转录起始或调节网络的复杂调节的模型)、酵母菌基因组数据库(酵母菌模式生物的基因组)、病毒生物信息学资源中心(经整理的数据库,其包含11个病毒家族的注释基因组数据)、seed平台(包括所有完整的微生物基因组和大多数部分的基因组,该平台使用子系统用于对微生物基因组进行注释)、wormbaseparasite(寄生物种)、ucsc疟疾基因组浏览器(导致疟疾的物种(恶性疟原虫(plasmodiumfalciparum)等)的基因组)、大鼠基因组数据库(褐家鼠(rattusnorvegicus)的基因组和表型数据)、integrall(致力于涉及抗生素抗性的整合子、细菌遗传元件的数据库)、vectorbase(niaid人病原体无脊椎动物载体的生物信息学资源中心)、ezgenome、有关原核生物(古核生物(archaea)和细菌)的人工策划的基因组项目的全面信息、genedb(顶复合器门原生动物(apicomplexanprotozoa)、动基体目原生动物(kinetoplastidprotozoa)、寄生蠕虫(parasitichelminths)、寄生物载体以及数种细菌和病毒)、eupathdb(真核病原体数据库资源,包含变形虫(amoeba)、真菌、疟原虫、锥虫等)、1000基因组项目(提供来自许多不同民族的多于一千名匿名参与者的基因组)、个人基因组项目(提供人基因组)。
其他数据库可包括个性化数据库,例如包含同一对象的健康和患病组织的遗传信息的数据库。这样的数据库可用于例如用于在对象中筛选治疗之后癌症的再现或监测治疗有效性的方法中。
在本发明的上下文中,术语“序列读取”或“读取”可互换使用,并且是指已通过测序确定了核苷酸序列的任何大小的特定核酸,并且其优选被分配给物种,优选映射至相应物种的基因组。在一个优选的实施方案中,将读取分类为特定的物种,例如对象和/或微生物,优选分类为特定微生物。在一个实施方案中,读取可通过其丰度被归一化。
在另一些实施方案中,本发明涉及用于在对象中诊断疾病状态或疾病(例如感染性疾病)的方法,其中实施根据本发明的用于在所述对象中确定疾病状态或疾病的方法。
在一个实施方案中,本发明提供了用于监测对象的感染状态的方法,优选地用于在治疗和针对治疗的响应期间监测对象的方法,其中实施根据本发明的用于确定所述对象的感染状态的方法。
这样的方法优选地涉及鉴定患有疾病的对象,优选地涉及筛选疾病,优选地涉及预防医学分析。在一个优选的实施方案中,这样的方法鉴定对象中微生物的出现与疾病的发生的相关性。
本发明优选地涉及一种方法,其中致病性条件的特征在于至少一种微生物,例如至少一种病毒、细菌、真菌或寄生生物体的核酸的异常的量,特别是致病的量。
任何微生物,优选其核酸序列已知的微生物,可被确定存在于对象中,以及可被确定为对象中疾病的病原体。可在对象中确定其存在的一些示例性微生物包括病毒、细菌、真菌和寄生物。一些示例性细菌包括但不限于:脑膜炎奈瑟菌(neisseriameningitis);肺炎链球菌(streptococcuspneumoniae);酿脓链球菌(streptococcuspyogenes);卡他莫拉菌(moraxellacatarrhalis);百日咳博德特菌(bordetellapertussis);金黄色葡萄球菌(staphylococcusaureus);破伤风梭菌(clostridiumtetani);白喉棒状杆菌(corynebacteriumdiphtheria);流感嗜血杆菌(haemophilusinfluenza);铜绿假单胞菌(pseudomonasaeruginosa);无乳链球菌(streptococcusagalactiae);沙眼衣原体(chlamydiatrachomatis);肺炎衣原体(chlamydiapneumoniae);幽门螺杆菌(helicobacterpylori);大肠杆菌(escherichiacoli);炭疽杆菌(bacillusanthracis);鼠疫耶尔森氏菌(yersiniapestis);表皮葡萄球菌(staphylococcusepidermis);产气荚膜梭菌(clostridiumperffingens);肉毒梭菌(clostridiumbotulinum);嗜肺军团菌(legionellapneumophila);伯氏考克斯氏体(coxiellabumetii);布鲁氏菌属(brucellaspp.),例如流产布鲁氏菌(b.abortus)、犬布鲁氏菌(b.canis)、马耳他布鲁氏菌(b.melitensis)、木鼠布鲁氏菌(b.neotomae)、绵羊布鲁氏菌(b.ovis)、猪布鲁氏菌(b.suis)、鳍脚类布鲁氏菌(b.pinnipediae);弗朗西斯氏菌属(francisellaspp.),例如新凶手弗朗西斯氏菌(f.novicida)、蜃楼弗朗西斯氏菌(f.philomiragia)、土拉热弗朗西斯氏菌(f.tularensis);淋病奈瑟菌(neisseriagonorrhoeae);苍白密螺旋体(treponemapallidum);杜克雷嗜血杆菌(haemophilusducreyi);粪肠球菌(enterococcusfaecalis);屎肠球菌(enterococcusfaecium);腐生葡萄球菌(staphylococcussaprophyticus);小肠结肠炎耶尔森氏菌(yersiniaenterocolitica);结核分支杆菌(mycobacteriumtuberculosis);立克次氏体属(rickettsiaspp.);单核细胞增多性李斯特菌(listeriamonocytogenes);霍乱弧菌(vibriocholera);伤寒沙门菌(salmonellatyphi);布氏疏螺旋体(borreliaburgdorferi);牙龈卟啉单胞菌(porphyromonasgingivalis);克雷伯菌属(klebsiellaspp.),肺炎克雷伯菌(klebsiellapneumoniae)。
一些示例性病毒包括但不限于:正黏病毒科(orthomyxoviridae),例如甲型、乙型或丙型流感病毒;副黏病毒科(paramyxoviridae)病毒,例如肺炎病毒(例如呼吸道合胞病毒(respiratorysyncytialvirus,rsv))、腮腺炎病毒属(rubulavirus)(例如腮腺炎病毒)、副黏病毒(例如副流感病毒)、偏肺病毒(metapneumovirus)和麻疹病毒(例如麻疹);痘病毒科(poxviridae),例如正痘病毒(例如,天花病毒,包括主天花病毒和次天花病毒);小rna病毒科(picomaviridae),例如肠道病毒(例如,脊髓灰质炎病毒,例如1型、2型和/或3型脊髓灰质炎病毒;ev71肠道病毒;柯萨奇a或b病毒)、鼻病毒、嗜肝rna病毒、心病毒和口蹄疫病毒;布尼亚病毒,例如正布尼亚病毒(例如加州脑炎病毒),白蛉病毒属(phlebovirus)(例如裂谷热病毒(riftvalleyfevervirus))或神经病毒(例如克里米亚-刚果出血热病毒);嗜肝rna病毒属(例如,甲型肝炎病毒(hepatitisavirus,hav)、乙型和丙型肝炎病毒);丝状病毒科(filoviridae)(例如埃博拉病毒(包括扎伊尔(zaire)、象牙海岸(ivorycoast)、雷斯顿(reston)或苏丹(sudan)埃博拉病毒)或马尔堡病毒);披膜病毒(例如风疹病毒(rubivirus)、甲病毒和动脉炎病毒,包括风疹病毒(rubellavirus));黄病毒(例如森林脑炎(tick-borneencephalitis,tbe)病毒、登革热(1、2、3或4型)病毒、黄热病毒、日本脑炎病毒、科萨努尔森林病毒(kyasanurforestvirus)、西尼罗河脑炎病毒(westnileencephalitisvirus)、圣路易斯脑炎病毒(st.louisencephalitisvirus)、俄罗斯春夏脑炎病毒(russianspring-summerencephalitisvirus)和波瓦生脑炎病毒(powassanencephalitisvirus));瘟病毒(例如牛病毒性腹泻(bvdv)、古典猪瘟(csfv)和边界病(bdv));嗜肝dna病毒(例如,乙型肝炎病毒、丙型肝炎病毒、丁型肝炎病毒、戊型肝炎病毒或庚型肝炎病毒);弹状病毒(rhabdovirus)(例如,狂犬病毒属(lyssavirus),狂犬病病毒和水泡性病毒(vsv));杯状病毒科(caliciviridae)(例如诺沃克病毒(诺如病毒)和诺沃克样病毒,例如夏威夷病毒(hawaiivirus)和雪山病毒(snowmountainvirus));冠状病毒(例如sars冠状病毒、禽传染性支气管炎(avianinfectiousbronchitis,ibv)病毒、小鼠肝炎病毒(mousehepatitisvirus,mhv)和猪传染性胃肠炎病毒(porcinetransmissiblegastroenteritisvirus,tgev));逆转录病毒(例如致癌病毒、慢病毒(例如hiv-1或hiv-2)或泡沫病毒(spumavirus));呼肠孤病毒(例如正呼肠孤病毒、轮状病毒、环状病毒和科罗拉多蜱传热症病毒(coltivirus));细小病毒(例如细小病毒b19);疱疹病毒(例如,人疱疹病毒,例如单纯疱疹病毒(herpessimplexvirus,hsv),例如1型和2型hsv,水痘带状疱疹病毒(varicella-zostervirus,vzv),eb病毒(epstein-barrvirus,ebv),巨细胞病毒(cytomegalovirus,cmv),人疱疹病毒6(hhv6),人疱疹病毒7(hhv7)和人疱疹病毒8(hhv8));乳多空病毒科(papovaviridae)(例如乳头瘤病毒和多瘤病毒,例如血清型1、2、4、5、6、8、11、13、16、18、31、33、35、39、41、42、47、51、57、58、63或65,优选来自血清型6、11、16和/或18中的一种或更多种);腺病毒,例如腺病毒血清型36(ad-36)。
一些示例性真菌包括但不限于:皮肤真菌(dermatophytre),包括絮状表皮癣菌(epidermophytonfloccusum)、头癣小孢子菌(microsporumaudouini)、犬小孢子菌(microsporumcanis)、扭曲小孢子菌(microsporumdistortum)、马小孢子菌(microsporumequinum)、石膏样小孢子菌(microsporumgypsum)、矮小小孢子菌(microsporumnanum)、同心毛藓菌(trichophytonconcentricum)、马毛藓菌(trichophytonequinum)、鸡毛藓菌(trichophytongallinae)、石膏样毛藓菌(trichophytongypseum)、麦格氏毛藓菌(trichophytonnaegnini)、须毛藓菌(trichophytonmentagrophytes)、昆克努毛藓菌(trichophytonquinckeanum)、红色毛藓菌(trichophytonrubrum)、许兰毛藓菌(trichophytonschoenleini)、断发毛藓菌(trichophytontonsurans)、疣状毛藓菌(trichophytonverrucosum)、疣状毛藓菌白色变种(t.verrucosumvar.album)、盘状变种(var.discoides)、赭黄变种(var.ochraceum)、紫色毛藓菌(trichophytonviolaceum)和/或蜜块状毛藓菌(trichophytonfaviforme);烟曲霉(aspergillusfumigatus)、黄曲霉(aspergillusflavus)、黑曲霉(aspergillusniger)、构巢曲霉(aspergillusnidulans)、土曲霉(aspergillusterreus)、聚多曲霉(aspergillussydowi)、黄曲菌(aspergillusflavatus)、灰绿曲霉(aspergillusglaucus)、头状芽生裂殖菌(blastoschizomycescapitatus)、白色念珠菌(candidaalbicans)、烯醇酶念珠菌(candidaenolase)、热带念珠菌(candidatropicalis)、光滑念珠菌(candidaglabrata)、克鲁斯念珠菌(candidakrusei)、近平滑念珠菌(candidaparapsilosis)、类星形念珠菌(candidastellatoidea)、克鲁斯念珠菌、帕拉克斯念珠菌(candidaparakwsei)、葡萄牙念珠菌(candidalusitaniae)、假热带念珠菌(candidapseudotropicalis)、季也蒙念珠菌(candidaguilliermondi)、卡氏枝孢霉(cladosporiumcarrionii)、粗球孢子菌(coccidioidesimmitis)、皮炎芽生菌(blastomycesdermatidis)、新型隐球菌(cryptococcusneoformans)、棒地霉(geotrichumclavatum)、夹膜组织胞浆菌(histoplasmacapsulatum)、微孢子虫(microsporidia)、脑胞内原虫属(encephalitozoonspp.)、肠间隔微孢子虫(septataintestinalis)和毕氏肠微孢子虫(enterocytozoonbieneusi);短粒虫属(brachiolaspp.)、微孢子虫属(microsporidiumspp.)、小孢子虫属(nosemaspp.)、匹里虫属(pleistophoraspp.)、气管普孢虫属(trachipleistophoraspp.)、条孢虫属(vittaformaspp.)、巴西芽生菌(paracoccidioidesbrasiliensis)、卡氏肺孢子虫(pneumocystiscarinii)、苜蓿腐酶(pythiumninsidiosum)、皮屑芽胞菌(pityrosporumovale)、酿酒酵母(sacharomycescerevisae)、布拉酵母(saccharomycesboulardii)、粟酒酵母(saccharomycespombe)、尖端赛多孢子菌(scedosporiumapiosperum)、申克氏孢子丝菌(sporothrixschenckii)、白色毛孢子菌(trichosporonbeigelii)、鼠弓形体(toxoplasmagondii)、马尔尼菲青霉菌(penicilliummameffei)、马拉色菌属(malasseziaspp.)、着色真菌属(fonsecaeaspp.)、王氏霉菌属(wangiellaspp.)、孢子丝菌属(sporothrixspp.)、蛙粪霉属(basidiobolusspp.)、耳霉属(conidiobolusspp.)、根霉菌属(rhizopusspp.)、毛霉属(mucorspp.)、犁头霉属(absidiaspp.)、被孢霉属(mortierellaspp.)、小克银汉霉属(cunninghamellaspp.)、瓶霉属(saksenaeaspp.)、链格孢菌属(alternariaspp.)、弯孢菌属(curvulariaspp.)、长蠕孢菌属(helminthosporiumspp.)、镰孢菌属(fusariumspp.)、曲霉菌属(aspergillusspp.)、青霉菌属(penicilliumspp.)、褐腐病菌属(monoliniaspp.)、丝核菌属(rhizoctoniaspp.)、拟青霉属(paecilomycesspp.)、皮司霉属(pithomycesspp.)和枝孢属(cladosporiumspp.)。
一些示例性的寄生物包括但不限于:疟原虫(plasmodium),例如恶性疟原虫(p.falciparum)、间日疟原虫(p.vivax)、三日疟原虫(p.malariae)和卵形疟原虫(p.ovale),以及来自鱼虱科(caligidae)的那些寄生物,特别是来自疮痂鱼虱属(lepeophtheirus)和鱼虱属(caligusgenera)的那些寄生物,例如海虱(sealice),例如鲑疮痂鱼虱(lepeophtheirussalmonis)和智利鱼虱(caligusrogercresseyi)。
在本发明的上下文中,术语“抗生素抗性”意指细菌对抗生素的杀伤或抑制生长的特性的易感性的丧失。其还涉及微生物对抗微生物药的抗性,所述抗微生物药最初对由该微生物引起的感染的治疗有效。抗性微生物,包括细菌、真菌、病毒和寄生物,能够承受抗微生物药的攻击,例如抗细菌药、抗真菌药、抗病毒药和抗疟疾药的攻击,以使得标准治疗变得无效并且感染继续存在。
根据本发明,术语“肿瘤”或“肿瘤疾病”是指细胞(被称为赘生性细胞、肿瘤发生性细胞或肿瘤细胞)的异常生长,优选形成肿胀或病灶。“肿瘤细胞”意指通过快速的不受控制的细胞增殖而生长并在引发新生长的刺激停止之后继续生长的异常细胞。肿瘤显示出结构组织和与正常组织的功能协调的部分或完全缺失,并且通常形成独特的组织团块,其可能是良性的、恶化前的或恶性的。
癌症(医学术语:恶性赘生物)是一类这样的疾病,其中一组细胞显示出不受控制的生长(分裂超出正常范围)、侵袭(侵入和破坏邻近组织)且有时转移(通过淋巴或血液扩散至身体的其他位置)。癌症的这三种恶性特征将它们与具有自限性且不会侵袭或转移的良性肿瘤区分开。大多数癌症形成肿瘤,但一些癌症(例如白血病)则不会。恶性(malignancy)、恶性赘生物和恶性肿瘤基本上与癌症同义。
赘生物是瘤形成(neoplasia)引起的异常组织团块。瘤形成(希腊语的新生长)是细胞的异常增殖。细胞的生长超过其周围的正常组织的生长并且与之不协调。即使在刺激停止后,生长仍以同样过度的方式持续存在。这通常会导致肿块或肿瘤。赘生物可以是良性的,恶化前的或恶性的。
根据本发明的“肿瘤的生长”或“肿瘤生长”涉及肿瘤增大其尺寸的趋势和/或肿瘤细胞增殖的趋势。
出于本发明的目的,术语“癌症”和“癌症疾病”与术语“肿瘤”和“肿瘤疾病”可互换使用。
癌症根据类似于肿瘤和因此被认为是肿瘤的起源的组织的细胞类型进行分类。这些分别是组织学和位置。
根据本发明的术语“癌症”包括上皮癌(carcinoma)、腺癌、母细胞瘤、白血病、精原细胞瘤、黑素瘤、畸胎瘤、淋巴瘤、神经母细胞瘤、神经胶质瘤、直肠癌、子宫内膜癌、肾癌、肾上腺癌、甲状腺癌、血癌、皮肤癌、脑癌、宫颈癌、肠癌、肝癌、结肠癌、胃癌、肠癌、头颈癌、胃肠癌、淋巴结癌、食管癌、结直肠癌、胰腺癌、耳鼻喉(ent)癌、乳腺癌、前列腺癌、子宫癌、卵巢癌和肺癌,及其转移。其实例是肺癌、乳腺癌、前列腺癌、结肠癌、肾细胞癌、宫颈癌,或上述癌症类型或肿瘤的转移。根据本发明的术语癌症还包括癌症转移和癌症复发。
根据本发明,“上皮癌”是来源于上皮细胞的恶性肿瘤。该组代表最常见的癌症,包括乳腺癌、前列腺癌、肺癌和结肠癌的常见形式。“腺癌”是源自腺组织的癌症。该组织也是被称为上皮组织的一大类组织的一部分。上皮组织包括皮肤、腺体以及内衬于身体的腔和器官的多种其他组织。在胚胎学上,上皮来自外胚层、内胚层和中胚层。被归类为腺癌的细胞并不一定必须是腺体的一部分,只要它们具有分泌特性即可。该形式的上皮癌可发生在一些包括人在内的高等哺乳动物中。良好分化的腺癌趋向于与其所来源的腺组织类似,而不良分化的腺癌则可能不是这样。通过对来自活检的细胞进行染色,病理学家将确定肿瘤是腺癌还是一些其他类型的癌症。由于腺体在体内普遍存在的性质,腺癌可在身体的许多组织中产生。尽管每种腺体可不分泌相同的物质,但只要细胞具有外分泌功能,它就可被认为是腺性的,并且因而它的恶性形式被命名为腺癌。只要有充足的时间,恶性腺癌就侵袭其他组织并且经常转移。卵巢腺癌是最常见的卵巢癌类型。其包括浆液性和黏液性腺癌、透明细胞腺癌和子宫内膜样腺癌。
“转移”意指癌细胞从其原始部位扩散到身体的另一部分。转移的形成是很复杂的过程,并且取决于恶性细胞从原发性肿瘤脱离,侵袭细胞外基质,渗透内皮基底膜以进入体腔和血管,以及然后在通过血液转运后浸润靶器官。最终,在靶位点的新肿瘤,即继发性肿瘤或转移性肿瘤的生长取决于血管生成。甚至在去除原发性肿瘤之后,也经常发生肿瘤转移,这是因为肿瘤细胞或组分可能保留并发展转移潜力。在一个实施方案中,根据本发明的术语“转移”涉及“远端转移”,其涉及远离原发性肿瘤和区域性淋巴结系统的转移。
继发性或转移性肿瘤的细胞与原始肿瘤中的细胞类似。这意味着,例如,如果乳腺癌转移至肝,则继发性肿瘤由异常乳腺细胞而不是异常肝细胞构成。肝中的肿瘤然后被称为转移性乳腺癌,而不是肝癌。
术语“循环肿瘤细胞”或“ctc”涉及已从原发性肿瘤或肿瘤转移瘤脱离并且在血流中循环的细胞。ctc可构成随后在不同组织中生长另外的肿瘤(转移)的种子。循环肿瘤细胞以约1至10个ctc/ml全血的频率存在于患有转移性疾病的患者中。已开发了研究方法来分离ctc。本领域中已描述了若干种研究方法来分离ctc,例如使用上皮细胞通常表达细胞黏附蛋白epcam(其在正常血细胞中不存在)的这一事实的技术。基于免疫磁珠的捕获涉及用已与磁性颗粒缀合的针对epcam的抗体处理血液试样,随后在磁场中分离标记的细胞。然后,将分离的细胞用针对另一上皮标志物细胞角蛋白和常见的白细胞标志物cd45的抗体染色,以使罕见的ctc与污染白细胞区分开来。这种稳健且半自动化的方法以平均产量为约1个ctc/ml且纯度为0.1%鉴定ctc(allard等,2004,clincancerres10:6897-6904)。用于分离ctc的第二种方法使用基于微流体的ctc捕获装置,其涉及使全血流过嵌入有80,000个微柱的室,所述微柱通过用针对epcam的抗体包被而变得具有功能性。ctc然后用针对细胞角蛋白或组织特异性标志物(例如前列腺癌中的psa或乳腺癌中的her2)的二抗进行染色,并且通过沿着三维坐标在多个平面中自动化扫描微柱来可视化。ctc芯片能够以50个细胞/ml的中值产量和1%至80%的纯度来鉴定患者中的细胞角蛋白化阳性循环肿瘤细胞(nagrath等,2007,nature450:1235-1239)。用于分离ctc的另一种司能性是使用来自veridex,llc(raritan,nj)的cellsearchtm循环肿瘤细胞(ctc)测试,其对血液管中的ctc进行捕获、鉴定和计数。cellsearchtm系统是美国食品和药物管理局(fda)批准的用于全血中ctc计数的方法,其基于免疫磁性标记和自动化数字显微术的组合。存在文献中描述的用于分离ctc的其他方法,所有这些方法均可以与本发明结合使用。
当个体再次受到过去影响其的病症影响时,出现复发或再现。例如,如果患者曾患有肿瘤疾病,已经接受所述疾病的成功治疗,并且再次发生所述疾病,则所述新发生的疾病可认为是复发或再现。然而,根据本发明,肿瘤疾病的复发或再现可以但不一定发生在原始肿瘤疾病的部位。因此,例如,如果患者曾患有乳腺肿瘤并且已接受了成功治疗,则复发或再现可为在与乳腺不同的部位处出现乳腺肿瘤或出现肿瘤。肿瘤的复发或再现还包括其中肿瘤出现在与原始肿瘤部位不同的部位以及出现在原始肿瘤部位的情况。优选地,患者已经接受治疗的原始肿瘤是原发性肿瘤,并且在与原始肿瘤部位不同的部位的肿瘤是继发性或转移性肿瘤。
“治疗”意指向对象施用本文中所述的化合物或组合物以预防或消除疾病,例如感染性疾病;并且还包括在对象中降低肿瘤的尺寸或肿瘤数量;阻止或减缓对象中的疾病;抑制或减缓对象中新疾病的发展;降低目前患有或曾患有疾病的对象中症状和/或复发的频率或严重程度;和/或延长(即增加)对象的寿命。特别地,术语“疾病的治疗”包括治愈、缩短持续时间、改善、预防、减缓或抑制进展或恶化,或者预防或延缓疾病或其症状的发作。
“处于风险中”意指对象,即患者,与一般群体相比,被鉴定为具有高于正常的发生疾病,特别是癌症的机会。此外,曾患有或目前患有疾病,特别是癌症的对象是具有提高的发生疾病的风险的对象,因为这样的对象可能继续发展疾病。目前患有或曾患有癌症的对象也具有提高的癌症转移风险。
在本发明的上下文中,术语例如“保护”、“预防”、“预防的”、“预防性的”或“保护性的”涉及对象中疾病发生和/或传播的预防或治疗或者这二者,并且特别地涉及使对象将发生疾病的机会最小化或延迟疾病的发生。例如,如上所述,处于肿瘤风险中的人将是用于治疗以预防肿瘤的候选者。
根据本发明的一个实施方案,一旦已经确定对象患有感染性疾病或其他疾病状态,则可向对象施用合适的治疗以治疗感染性疾病或其他疾病状态。这些治疗,包括抗生素和抗癌剂,在本领域中是公知的,并且最终将给予对象的合适治疗将由治疗医师确定。
在一个实施方案中,本发明还涉及用于实施根据本发明的方法的装置,其中以下由装置的中央处理单元计算:将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息。在一个实施方案中,本发明还涉及用于实施根据本发明的方法的装置,其中以下由装置的中央处理单元计算:随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目。在一个实施方案中,本发明还涉及用于实施根据本发明的方法的装置,其中以下由装置的中央处理单元计算:基于映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目,在对象中发现映射至特定微生物的所比较的序列读取的概率的显著性评分。在一个实施方案中,中央处理单元是现场可编程门阵列(field-programmablegatearray,fpga)。在一个优选的实施方案中,装置实施一个或更多个或所有前述计算。在一个实施方案中,本发明还涉及可实施一个或更多个或所有与在对象中确定疾病状态的存在相关的计算的装置。
因此,本发明提供了完整的诊断工作流,其用于基于核酸(例如游离循环dna)的无偏序列分析来确定生物样品中微生物或疾病状态的存在。在不知道可疑微生物或疾病状态的情况下,该方法有利地提供了数据驱动的诊断,不需要特定的引物设计,并且提供了在单个测定中检测多种病毒、细菌、真菌和寄生微生物的机会。
本发明的方法优选不限于特定微生物的确定。在一个实施方案中,本发明方法确定对象中所有微生物的存在,优选与疾病状态(例如感染)相关的所有微生物的存在。本发明的方法还优选地不限于在对象中确定特定的癌症类型,而是可确定多于一种的癌症类型以及癌症亚型的存在。在一个优选的实施方案中,对象中癌症的不同类型和/或亚型在其遗传物质中具有不同的突变,以使得可根据本发明的方法确定对象中一种或更多种癌症类型和/或癌症亚型的存在。
因此,本发明提供了用于在短时间内鉴定对象中感染或其他疾病状态的原因的有用方法,以使得可在短时间内选择用于所鉴定的感染或其他疾病状态的合适治疗。
因此,本发明的方法对于临床试样中数据驱动的微生物的鉴定,对于监测对象的微生物负荷以及针对靶向治疗的响应和补充标准临床微生物学可以是非常有用的。本发明的方法对于临床试样中数据驱动的肿瘤细胞存在的鉴定,对于监测对象的肿瘤细胞负荷以及针对靶向治疗的响应和补充标准临床肿瘤学也可以是非常有用的。
通过以下附图和实施例详细描述本发明,这些附图和实施例仅用于举例说明的目的并且不意在限制。由于描述和实施例,技术人员可得到同样包含在本发明中的另外的实施方案。
附图说明
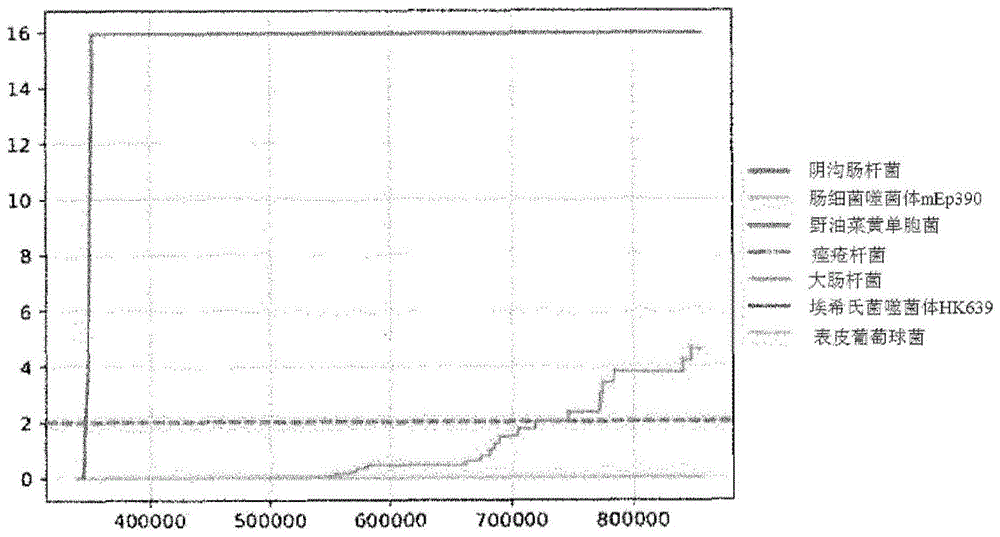
图1示出了针对7种不同微生物,患者s9的完整测试运行(对于被标记为显著的微生物,测试未停止)。还绘制了水平虚线,指示统计相关性阈值。
图2示出了针对4种不同微生物,患者s11的完整测试运行(对于被标记为显著的微生物,测试未停止)。还绘制了水平虚线,指示统计相关性阈值。
图3示出了针对5种不同微生物,患者s60的完整测试运行(对于被标记为显著的微生物,测试未停止)。还绘制了水平虚线,指示统计相关性阈值。
实施例
本文中使用的技术和方法在本文中进行了描述或者以本身已知的方式并且如例如sambrook等,molecularcloning:alaboratorymanual,2ndedition(1989)coldspringharborlaboratorypress,coldspringharbor,n.y中所述实施。除非特别地指出,否则包括试剂盒和试剂的使用的所有方法均根据制造商的信息进行。
实施例1
从被怀疑患有感染性疾病的人对象中获得生物样品,即血浆。使用下一代测序法对样品中的核酸进行测序,产生多个序列读取。存储该数据,然后如下进行分析。
将个体序列读取与包含人和多种微生物二者的遗传信息的一个或更多个数据库进行比较,以使得如果可能的话每个读取均映射至特定微生物或人基因组。所述映射实时提供了映射至特定微生物的读取的总数和可映射至物种(即特定微生物、人基因组以及任何其他微生物)的读取的总数。因此,在诊断程序期间的每个时间点处均已知归因于特定微生物或人对象的读取的数目。
该信息允许产生计数向量c:cm,...,cl;m=1,....l,其在诊断期间的任意但固定的时间点处保持样品/患者j中针对每个物种m的读取的数目。在患者j的诊断期间,cm随时间推移变化,同时新的读取映射至物种。此外,随着新微生物物种被鉴定,c可增长。首先,初始化一个空向量,并在该方法的运行时间期间生成一个动态向量。c描述了当前诊断的患者的微生物负荷。为了确定其负荷具有异常丰度的那些微生物,如下计算在给定时间处患者j中该特定微生物负荷的逆累积密度函数(cumulativedensityfunction,cdf):
其中cm是在当前时间时患者j中针对物种m测量的读取的数目,并且n是总的来说能够被映射的读取的数目(微生物和宿主)。pm描述了实时计算的发现概率,其表示检测出针对物种m的读取的概率。
与常规测试形成对比,这不是终点测试,而是在顺序测试的框架中运行。因此,通过顺序测试方法,可在测试运行时而不是测试完成之后获得所有必要且重要的信息。这提供了下一代测序领域中感染诊断的新方式和测试程序的新方式。所提供的信息是p值,它描述了映射至某个物种的核酸的当前量是否被认为是不正常的,因此,考虑到针对该物种的发现概率和当前运行测试设置,其会达到非常低的p值。
该方法允许定义新的特征变量,例如“微生物信号/次事件”。这些变量直接取决于微生物变得统计学相关的次数,因此,新的变量是特别重要的。可能的特征变量是“微生物读取/秒”或“微生物读取/人读取”。对于每个对象和每种微生物,这样的变量均可计算,并且因此将提供对每个所分析样品的感染严重程度的更深入的了解。此外,由于这些特征变量的技术独立性,这样的变量将能够比较用不同技术测序的样品。
实施例2
对来自从对象s9获得的血浆生物样品的核酸进行测序,以使得根据本发明计算基于映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目的在对象中发现映射至特定微生物的所比较的序列读取的概率。结果示于图1中。
图1示出了在同一时间,针对7种不同微生物的完整测试序列(测试未由于某些微生物的显著性而中断或中止)。还示出了水平的红色虚线,其表示统计阈值,在认为微生物与引起感染“相关”之前必须超过该统计阈值。同样清楚的是,表示微生物阴沟肠杆菌(enterobactercloacae)的蓝色线仅在产生数据的片刻之后就超过统计阈值,以使得对于该微生物,可在仅片刻之后终止测试。属于细菌大肠杆菌的紫色线示出了值的缓慢增加,但直到500k读取之后才超过表示相关的显著性水平,表明其与其他微生物是污染物或共生微生物。
实施例3
对来自从对象s11获得的血浆生物样品的核酸进行测序,以使得根据本发明计算基于映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目的在对象中发现映射至特定微生物的所比较的序列读取的概率。结果示于图2中。
与图1类似,图2示出了单一细菌(在此为肺炎克雷伯菌(k.pneumoniae)(绿色))与疾病状态相关(即为感染的病原体)的概率迅速增加。注意,检测到了痤疮杆菌(cutibacteriumache),其是生活在人皮肤上的细菌,但是该细菌成为感染的病原体的相关性/概率为零。这表明该方法如预期地,滤除了共生物种。相比之下,大肠杆菌的相关性在350k读取的时间范围内上升至显著性阈值。尽管未示出相关,但这可表明患者处于发生由大肠杆菌引起的继发性感染的危险中。
这表明该方法产生了当前“基于终点”的测试可能无法提供的信息。因此,该方法提供了指示临床医生在感染为实际临床相关的之前就针对感染采取行动的数据。本文中所述方法的另一个优点是能够检测由多种微生物引起的感染并进一步划分哪些微生物是主要的病原体。
实施例4
对来自从对象s60获得的血浆生物样品的核酸进行测序,以使得根据本发明计算基于映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目的在对象中发现映射至特定微生物的所比较的序列读取的概率。结果示于图3中。
如图3中清楚所示地,主要感染原是脆弱拟杆菌(b.fragilis),因为绿色线刚好在方法开始时超过相关性阈值。但是,在一些事件(读取被分析)之后,由分别为大肠杆菌和金黄色葡萄球菌(s.aureus)的橙色线和紫色线表示的另外两种细菌在相关性阈值之上有了显著飞跃,表明这两种细菌也促进对象的脓毒症。
将该结果与基于对所有三种细菌的常规测试的传统结局进行比较,结果将看起来是相同的。每种微生物被分配了或多或少相同的相关性。然而,使用本文中所述的方法,可清楚地鉴定主要病原体,并且通过客观使用特征变量(例如“事件/次”),鉴定主要病原体以及其他促进感染的微生物。
上图中的轴始终是用公式1计算的p值的对数和所分析的读取的数目。当然,可改变此轴上所示的单位。在此,完全有必要的是,通过新单位,读取的唯一排序是可能的。例如,这可能是生成读取的顺序或将它们与数据库进行比较的时间。使用上述方法,可计算上述特征变量,例如特定微生物和患者的“直至相关的读取/次事件”。这些变量可用于比较患有同一微生物的不同患者。此外,可通过比较同一患者中不同微生物的变量来鉴定主要病原体。
假设实际感染的范围是例如通过“读取/次事件”测量的某个区间,例如[x-y]。然后,污染物和共生物将示出在该“感染区间”的边界的外部上方。因此,使用这些感染区间的统计分析足以鉴定感染并评估所鉴定的微生物的相关性。此外,通过这些区间来评估感染的严重程度。这是使用等待时间分析(waitingtimeanalysis)的统计学框架来完成的。在大多数情况下,等待时间分析是使用指数函数进行的。因此,假设描述“特征感染变量”的变量是根据指数随机变量分布的:
x~exp(λ)[2]
并且假设某一微生物的等待时间在500至1000个读取之间,则λ=1/500和λ=1/1000。由于我们对p(500<x<1000)的概率感兴趣,因此我们计算p(x<1000)-p(x≤500)。这描述了未患有感染的概率。由于我们想要比此更快的区间,因此我们计算p(x≤500)。现在,如果第500个读取又是微生物读取,我们要做的就是计算p(x>500)=e-500λ≈0.36。因此,假定该特定物种的区间为500至1000,则很可能在宿主的500个读取之后观察到微生物读取。但是,如果我们仅在10个读取之后就观察到第二微生物读取,则我们计算p(x>10)=e-100λ≈0.98,因为我们在10个信号(所比较的读取)之后观察到微生物读取,我们对p(x≤10)感兴趣,并且因此1-p(x>10)=0.019。因此,极不可能在10个信号之后检测到微生物,因此,如果在10个信号之后检测到微生物,则需要将其报告给临床医生。
在感染性疾病诊断或一般诊断中,没有描述在给定一组事件的情况下将概率与固定但任意的事件量结合以及所得的等待时间分析这两种方法。通常来说,如果可将数据生成分为不同的通道或块,我们可再次将测试并行化到每个单独的通道(即,分别测试每个通道并作为单独的实验处理每个通道),并且因此使产生结果的时间最小化。同样,这使用终点测试也是不可能的,意味着与基于终点的测试形成对比,本文中所述的方法可朝更高的通量扩展。
本发明特别地提供以下内容:
1.用于在对象中确定微生物的存在的方法,其包括:
(a)对从所述对象获得的生物样品中存在的核酸进行测序以获得多个核酸序列读取;
(b)将步骤(a)中获得的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至所述一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息;以及
(c)随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目。
2.用于在对象中确定微生物的存在的方法,其包括:
(a)将序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至所述一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息,其中所述序列读取是通过对从所述对象获得的生物样品中存在的核酸进行测序而获得的;以及
(b)随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目。
3.根据条款1或2所述的方法,其中所述方法还包括基于映射至所述特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目,计算在所述对象中发现映射至所述特定微生物的所比较的序列读取的概率的显著性评分。
4.根据条款3所述的方法,其中当针对所述特定微生物的评分达到或超过阈值时,确定所述特定微生物存在于所述对象中。
5.根据条款3所述的方法,其中当针对所述特定微生物的评分达到或超过阈值时,确定所述特定微生物与在所述对象中引起疾病相关。
6.根据条款5所述的方法,其中当针对所述特定微生物的评分在少数序列读取就超过阈值时,由于所述微生物的存在而引起的疾病被认为是严重的。
7.用于在对象中确定疾病状态的存在的方法,其包括:
(a)对从所述对象获得的生物样品中存在的核酸进行测序以获得多个核酸序列读取;
(b)将步骤(a)中获得的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至对照对象,所述一个或更多个数据库包含来自同一物种的所述对照对象的遗传信息;以及
(c)随时间推移确定映射至和未映射至所述对照对象的所比较的序列读取的数目。
8.根据条款7所述的方法,其中所述方法还包括基于未映射至所述对照对象的所比较的序列读取的数目和映射至所述对照对象的所比较的序列读取的数目,计算在所述对象中发现未映射至所述对照对象的所比较的序列读取的概率的显著性评分。
9.根据条款8所述的方法,其中当所述评分达到或超过阈值时,确定所述疾病状态存在于所述对象中。
10.根据条款7至9中任一项所述的方法,其中所述疾病状态是癌症。
11.根据条款10所述的方法,其中所述癌症是由遗传异常引起的。
12.根据条款7至9中任一项所述的方法,其中所述疾病状态是由微生物引起的感染。
13.根据条款12所述的方法,其中所述微生物是病毒、细菌、真菌或寄生物。
14.根据前述条款中任一项所述的方法,其中所述生物样品选自全血、血清、血浆、羊水、滑液、液体、组织或细胞涂片、组织或细胞拭子、尿、组织、痰、粪便、胃肠道分泌物、淋巴液和灌洗液。
15.根据前述条款中任一项所述的方法,其中所述对象是脊椎动物,优选哺乳动物,例如人、狗、猫、猪、马、牛、绵羊、山羊、小鼠或大鼠。
16.根据条款15所述的方法,其中所述对象是人。
17.根据前述条款中任一项所述的方法,其中所述测序是通过分子高通量序列分析进行的。
18.根据前述条款中任一项所述的方法,其中当确定所述特定微生物或所述疾病状态存在于所述对象中时,所述方法还包括向所述对象施用已知治疗由所述特定微生物引起的疾病或所述疾病状态的药物活性化合物。
19.用于在对象中诊断由微生物引起的感染性疾病的方法,其包括:
(a)对从所述对象获得的生物样品中存在的核酸进行测序以获得多个核酸序列读取;
(b)将步骤(a)中获得的序列读取与一个或更多个数据库进行比较以确定所比较的序列读取是否映射至所述一个或更多个数据库中包含的物种,所述一个或更多个数据库包含来自同一物种的对照对象的遗传信息和来自多种微生物的遗传信息;
(c)随时间推移确定映射至特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目;以及
(d)基于映射至所述特定微生物的所比较的序列读取的数目和映射至物种的所比较的序列读取的数目,计算在所述对象中发现映射至所述特定微生物的所比较的序列读取的概率的显著性评分,
其中当针对所述特定微生物的评分达到或超过阈值时,确定所述特定微生物引起所述感染性疾病。
20.存储程序代码的计算机可读存储介质,其包含指令,所述指令当由处理器执行时实施根据条款1至19中任一项所述的方法。
21.计算机系统,其包含被配置为实施根据条款1至19中任一项所述的方法的处理器。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于CRISPR‑Cas9系统的基因编辑载体及其应用的制造方法与工艺
- 一种基于CRISPR/Cas9技术制备水稻OsPIL15突变体的方法及应用与流程
- 检测黄曲霉毒素M1的核酸适配体、传感器、试剂盒及应用的制造方法与工艺
- 一组肌酸激酶同工酶核酸适配体及其应用的制造方法与工艺
- 用于抑制KRAS靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 用于抑制EIF4E靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 用于抑制VEGFA靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 一种新型向导RNA表达盒及在CRISPR/Cas系统中的应用的制造方法与工艺
- 一种具备双单链末端PCR产物的获得方法及其检测方法与流程
- 一种通过生物技术扩增脱氧胸苷三磷酸的方法与流程