用于测定患有增生疾病的患者对使用靶向PD1/PD-L1路径的组分的药剂的治疗的敏感性的方法与流程
本发明涉及一种用于测定患有如癌症的增生疾病的患者对使用特定类型的药剂的治疗的敏感性的方法。其进一步包含开发基于测定的针对所选患者的治疗方案,用于进行测定的试剂盒以及经编程以进行测定的计算机。
背景技术:
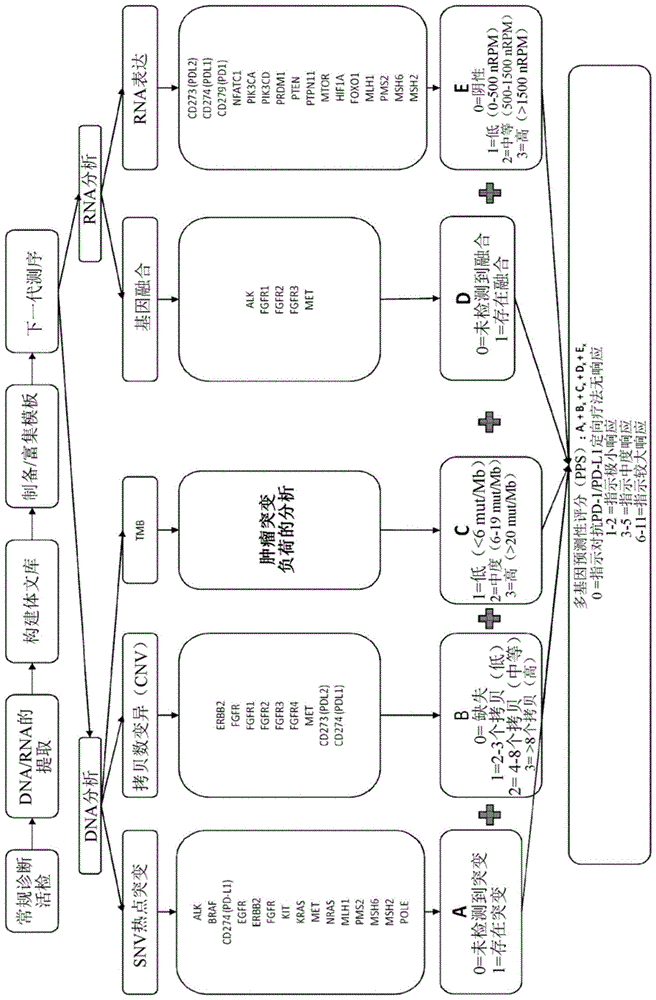
:抗pd-1/pd-l1定向免疫疗法已变成免疫疗法中所用的最重要的一组药剂之一。pd-1/pd-l1路径通常参与促进耐受性并防止在慢性炎症环境中发生组织损伤。程序性死亡1(pd-1)以及其配体,pd-l1和pd-l2递送调节t细胞活化、耐受性与免疫病理学之间的平衡的抑制信号。pd-1程序性死亡-配体1(pd-l1)为免疫系统调节期间结合于程序性死亡-1受体(pd-1)的跨膜蛋白。这一pd-1/pd-l1相互作用通过抑制t细胞作用保护正常细胞不被免疫识别,由此防止发生免疫介导的组织损伤。在对抗癌症中利用免疫系统已变成主要关注话题。用于治疗癌症的免疫疗法是由总体性地和非特异性地刺激免疫系统的疗法快速进化成更有针对性的方法的领域。pd-1/pd-l1路径已成为免疫疗法的强有效目标。已显示有一系列癌症类型会表达pd-l1,其结合于由免疫细胞表达的pd-1,产生免疫抑制作用,使得这些癌症逃避肿瘤破坏。pd-1/pd-l1相互作用抑制t细胞活化并增强调节性t细胞(t-reg)增殖,其进一步抑制对抗肿瘤的效应免疫响应。这与正常细胞所使用的避免免疫识别的方法相似。因此靶向pd-1/pd-l1已成为免疫疗法定向疗法的新型和强有效方法。用定位在pd-1和pd-l1处的治疗抗体靶向pd-1/pd-l1路径已成为显示免疫逃避特征的那些癌症类型中的强有效疗法。用针对pd-1或pd-l1的治疗抗体(抗pd-l1或抗pd-1药剂)干扰pd-1/pd-l1路径使得效应免疫响应得到修复,伴随针对肿瘤的t细胞得到优先活化。已知包括黑素瘤、肾细胞癌、头颈部的肺癌、胃肠道恶性疾病、卵巢癌、血液科恶性疾病在内的一系列癌症类会表达pd-l1,引起免疫逃避。已显示抗pd-l1和抗pd-1疗法在多种这些肿瘤类型中诱发强烈的临床响应,例如在黑素瘤中,呈20-40%,并且在晚期非小细胞肺癌(nsclc)中,呈33-50%。这些抗体中有多种抗体,例如抗pd-1定向药剂纳武单抗(nivolumab)和派立珠单抗(pembrolizumab)现已得到fda批准用于治疗转移性nsclc和晚期黑素瘤。有九种靶向pd-1/pd-l1路径的开发药物,并且医药公司的现有实践在于独立地开发出作为对抗pd-1/抗pd-l1定向疗法的响应的预测因子的抗pd-l1免疫组织化学(ihc)诊断测定。这些pd-1/pd-l1定向疗法包括派立珠单抗、阿特珠单抗(atezolizumab)、艾维路单抗(avelumab)、纳武单抗、度伐单抗(durvalumab)、pdr-001、bgb-a317、regw2810、shr-1210(下文中的表1)。领先的生物制药公司在石蜡包埋的福马林固定的诊断活检体和切除组织/样品(pwet)上均已选择免疫组织化学方法,以产生针对抗pd-1/pd-l1定向疗法的伴随诊断。所有这些测试均涉及使用标准免疫组织化学测定方法以及酶联色原体检测系统施用单克隆抗体,其针对施用于组织切片的pd-l1而产生。随后通过病理学家进行微观检查以测定表达pd-l1的细胞比例来人工地评定细胞的免疫组织化学染色(针对pd-l的部分或完全表面膜染色)。这随后报告肿瘤比例评分。一些测定仅评定pd-l1的肿瘤细胞表达,而其它测定评定肿瘤细胞和相关肿瘤内和肿瘤周围免疫细胞浸润(ic)中的pd-l1的表达。肿瘤比例评分被定义为相对于在样品中存在的所有活肿瘤细胞(阳性和阴性),显示部分或完全膜染色的活肿瘤细胞的百分比(≥1+)。评定针对pd-l1的肿瘤比例评分或肿瘤比例评分和免疫细胞浸润评分的伴随诊断测定的代表性实例示于下文(下文中的表2)。这些测试已由达科公司(dako)和本塔纳公司(ventana)开发,其强调所述两种类型的方法。存在与现有的抗pd-1/pd-l1定向抗癌免疫疗法的基于ihcpd-l1的伴随诊断相关的主要问题。举例来说,到目前为止,每种治疗药物已被批准与使用不同pd-l1或pd-1抗体的单独伴随诊断测试结合。抗体在其敏感性和特异性方面可有极大不同。因此疗法与诊断的模型存在用于在临床上进行测试和决策的复杂挑战。此外,每个测定的目的已由临床经验形成。因此,用作伴随派立珠单抗的晚期nsclc试验中的患者富集的纳入标准的pd-l1ihc22c3pharmdx需要在这一适应症中临床使用所述药物,而在同一患者群中进行回顾性地评估的pd-l1ihc28-8pharmdx用于告知如由生物标记阳性界定的不同患者子组的风险-效益评定。本发明测定不包括pd-1路径中也相关的pd-l2配体的分析。与伴随现有pd-l1ihc测定的问题相交织,在不存在pd-1和pd-l2下评估pd-l1的分析会损害pd1/pd-l1/pd-l2信号传导网络的综合评定,其降低了精确鉴别可能会对抗pd-1/pd-l1定向疗法作出响应的患者的准确性。类似地,pd-1测试,就其自身而言,缺乏pd-l1和pd-l2分析所提供的预测性信息。除了pd-1和pd-l1之外,pd-l1信号传导轴还涉及其它主要组分,其已显示为对抗pd-1/pd-l1/pd-l2定向免疫治疗剂的响应的预测因子,包括增加的表达水平的nfatc1、pik3ca、pik3cd、prdm1、pten、ptpn11、mtor、hif1a、foxo1。使用常规ihc不可能分析所述广泛范围的异常基因表达。值得注意的是,宽范围的致癌突变也已显示为对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的主要预测因子。这些包括致癌基因和产生融合基因的致癌染色体重组事件中的热点突变。此外,ihc方法无法用于将所述宽范围的致癌基因事件评定为对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的预测性生物标记。pd-l1和pd-l2的异常过表达还可由于基因扩增而发生。基于ihc的方法不能够检测pd-l1和pd-l2配体的基因扩增。由于多种致癌基因可经历预测对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的扩增,因此使用ihc方法不可检测这些致癌基因。此外,pd-l1的编码序列中的突变为对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的预测因子,但无法被ihc检测到。pd-l1、pd-l2和pd-1表达的细胞、空间和时间不均匀性结合上文所论述的问题均促成临床上这些生物标记的较差预测准确性(即,缺乏阳性和阴性预测值)。此外,已显示经针对pd-l1、pd-l2和pd-1的ihc评分为阳性的肿瘤比例未能对抗pd-1/pd-l1/pd-l2定向免疫疗法作出响应,并且同样地,肿瘤活检体在pd-l1蛋白质表达方面呈pd-l1阳性的患者子组实际上几乎未实现临床益处。这再次表明,基于ihc的测定为不适当的,会引起产生错报和误报结果。pd-1/pd-l1/pd-l2免疫调节路径的综合分析需要分析参与抗肿瘤免疫响应的肿瘤中的所有细胞组分。这包括肿瘤细胞、免疫t细胞和抗原呈递细胞(aps),后者替代地称为巨噬细胞或树突状细胞。然而,大多数可利用的测定仅评定肿瘤细胞。pd-l1免疫染色的个别病理学家进行的微观检查会根据其训练和经验而有所变化。因此在针对pd-l1的肿瘤比例评分的产生过程中存在严重的观察者间变化。此外,标记指数的评定极其困难,如单一病理学家的高内变化水平中所突显。现有的基于ihc的测定使用不同抗体克隆,其在敏感性和特异性方面有所不同,并且由不同病理学家进行解释,所述病理学家在解释肿瘤比例评分和染色强度方面有所不同。这还适用于针对pd-l1表达的免疫细胞的评定。这会被与不同肿瘤类型相关的不同组织架构进一步混淆。综合起来,这些问题使得实验室中的标准化极为困难。在尝试开发ki67标记指数方案的早先研究中突显病理学家产生准确的ihc生物标记的标记指数的问题。值得注意的是,自动化图像分析也未能解决这一问题。此外,抗体克隆的选择,pd-l1染色的最佳截止点标准化存在问题并且尚未得到确立。最后,本质上,基于ihcpd-l1的测试在根本上为半定量的,并且因此缺乏定量测定所提供的精确度。使用下一代测序技术和t细胞或b细胞组库的多样性分析(免疫组学(immunome))的全基因组突变分析(突变组学(mutanome))由于产生作为用于鉴别预测性生物标记的潜在策略的关注而得到报告(y.iwai等人生医科学杂志(journalofbiomedicalscience)(2017)24:26doi10.1186/sl2929-017-0329-9)。然而,已意识到生物标记的选择至关重要并且根据癌症类型,个别生物标记并非始终对应于高响应。技术实现要素:根据本发明,提供一种用于测定患有增生疾病的患者对使用靶向pd1/pd-l1路径的组分的药剂的治疗的敏感性的方法,所述方法包含在来自所述患者的肿瘤样品中测定至少三种生物标记,所述至少三种生物标记选自下文中的表3a-3d中所阐述的群组;并使多于一种所述生物标记的存在关联为对所述药剂的敏感性指标。如本文所用,术语“生物标记”是指任何分子、基因、序列突变或特征,如增加或降低的基因表达,其指示pd1/pdl1路径中的异常。这些可以包括基因序列中的突变,尤其已知会产生肿瘤后果、基因的拷贝数变异、异常基因融合或增加或降低的rna表达的‘热点’突变。靶向pd1/pd-l1路径的组分的药剂将宜为靶向或结合于pd1、pd-l1或pd-l2蛋白质的药剂,但还可设想靶向所述蛋白质,例如,编码那些蛋白质的dna或rna的表达的药剂。确切地说,可用作供用于本发明方法中的生物标记的基因和与癌症风险相关的变化阐述于下文中的表3a-3d和图1中。在一个特定实施例中,本发明方法将涉及测定表1中的至少5种,例如至少8种,如至少10种生物标记,并且优选地,测定表1中的所有生物标记。确切地说,所用生物标记数越大,将鉴别到失调基因的概率越大,以使得避免发生漏报(即,其中遗漏易患病患者)。在一个特定实施例中,所检测到的生物标记中的至少一种与pd-1/pd-l1路径直接相关,并且因此为选自cd279(pd1)、cd274(pd1)或cd273(pd2)的基因的生物标记。在一个特定实施例中,在一起评定pd-l1和pd-1,以提供pd-1/pd-l1信号轴的更强有效的评定结果。在另一个具体实施例中,所述方法测量pd-l1和pd-l2基因扩增(拷贝数变异体;cnv),其与mrna过表达相联系,并可表示预测对pd-1/pd-l1抑制剂的响应的较可靠的参数。在一个特定实施例中,本发明方法分析和整合参与肿瘤-免疫细胞相互作用中的所有细胞群中的pd-1和pd-l1表达,所述细胞群包括肿瘤细胞、免疫细胞和抗原呈递细胞(apc)。在一个特定实施例中,所检测到的基因中的至少一种不与pd1/pdl-1路径直接相关,但为表3a-3d中所列的生物标记,其不为cd279(pd1)、cd274(pd1)或cd273(pd2)。通过选择与不同功能相关的一系列生物标记,申请者已了解对靶向免疫路径,并且确切地说,pd-1/pd-l1的治疗的敏感性的较好指示。其它基因中的突变,确切地说致癌突变,可能产生所谓的‘新抗原’。新抗原呈突变形式并且确切地说,为癌症特异性抗原,如果免疫系统有效并且未经历抑制,那么其可引起对抗癌细胞的t细胞活化。因此,在存在新抗原的情况下,患者对作用于如pd-1/pd-l1路径的免疫路径的药剂可显示更高效和持久的响应。在另一实施例中,根据本发明方法测量的另一生物标记为肿瘤突变负荷(tmb)。不同于蛋白质类生物标记,tmb为根据肿瘤基因组的编码面积的总突变数的定量量度。由于仅一部分体细胞突变会产生新抗原,因此测量特定编码面积中的总体细胞突变数(tmb)充当新抗原负荷的代表。可使用外显子组测序,确切地说,使用下一代测序,尤其覆盖1.7mb编码区的411个基因的下一代测序来测量tmb。从所检测到的所有变异体(包括插入缺失、取代等)中,从分析去除所有可能的生殖系多态性和经预测的致癌驱动基因。进行后者以防止出现对已知癌症基因测序的确定偏差。肿瘤突变负荷随后被计算为每mb测序dna的突变数(mut/mb)。tmb的分析为定量和定性的,并以度量值(mut/mb)以及状态报告。高状态归类为>20mut/mb,中等状态归类为6-19mut/mb,并且低状态归类为<6mut/mb。已意识到,tmb为对抗pd-1/pd-l1/pd-l2检查点抑制剂的响应的预测因子,并且因此其将提供适用于本发明方法的另外生物标记。可由所述药剂治疗的增生疾病包括癌症,确切地说,实体癌,如肾上腺癌、肛门癌、基底细胞和鳞状细胞皮肤癌、胆管癌、膀胱癌、骨癌、脑肿瘤和脊髓肿瘤、乳癌、原发灶不明癌、子宫颈癌、结肠直肠癌、子宫内膜癌、食道癌、尤文氏肉瘤(ewing'ssarcoma)、眼癌、胆囊癌、胃肠道类癌、胃肠道基质肿瘤(gist)、肾癌、喉和下咽癌、肝癌、肺癌、肺类癌肿瘤、恶性间皮瘤、黑素瘤、默克细胞皮肤癌(merkelcellskincancer)、鼻腔和副鼻窦癌、鼻咽癌、成神经细胞瘤、非小细胞肺癌、口腔和口咽癌、骨肉瘤、卵巢癌、胰腺癌、阴茎癌、垂体瘤、前列腺癌、成视网膜细胞瘤、横纹肌肉瘤、唾液腺癌、皮肤癌、小细胞肺癌、小肠癌、软组织肉瘤、胃癌、睾丸癌、胸腺癌、甲状腺癌、子宫肉瘤、阴道癌、外阴癌。用于测定特定生物标记的方法将视生物标记的性质而变化,并将为所属领域中所通常理解的。在生物标记基因表达蛋白质或肽,并且确切地说,变异体蛋白质或肽的情况下,可使用如elisa的免疫测定技术来鉴别这些基因。然而,在一个特定实施例中,使用dna或rna分析或其组合来鉴别生物标记。在一个特定实施例中,由患有癌症的患者获得肿瘤活检样品,如常规诊断pwet样品,并从其中提取核酸(dna和/或rna)。随后使用常规方法,例如如下文所概述的方法将这用于构建文库。根据需要富集文库并随后用作富集模板,同样使用如下文所概述的常规方法。随后使用半导体下一代测序技术,如可购自oncologicauk有限公司的技术,来对其进行分析。在一个特定实施例中,本发明方法使用经靶向的半导体测序以覆盖所关注生物标记的整个编码区。扩增子已针对序列覆盖冗余被设计成重叠的,并能够扩增由常规诊断pwet样品获得的片段化dna模板。因此所述方法能够鉴别多种可行的基因变异体,包括点突变、缺失、重复和插入。因此,在一个特定实施例中,本发明方法利用下一代测序技术以定量地测量来自样品,如取自常规的福马林固定的石蜡包埋的肿瘤样品的单一10μm切片的pd-1和pd-l1rna表达水平。因此,可以使用仅少量(<10ng)pwet物质来进行本发明方法,并其可经优化用于分析降解的dna/rna。所述方法可用于提供定量测试,其产生比免疫组织化学(ihc)要准确得多的方法以测定最可能对抗pd-1和抗pd-l1定向疗法作出响应的那些患者。通过避免使用抗体,与不同抗体克隆的敏感性和特异性相关的问题得到避免。一些生物标记可最容易通过基因表达的分析,例如使用rna转录物的定量测量得到鉴别。因此,在一个特定实施例中,所述方法包括分析rna水平的步骤,其中与野生型相比的表达水平的变化指示pd-1/pd-l1路径。可以此方式鉴别的生物标记列于下表3a和图1中。确切地说,在其中涉及所述路径的肿瘤中,可从cd273(pd-l2)、cd274(pd-l1)和cd279(pd-1)中检测到升高的rna水平。在一个特定实施例中,进行跨pd-l1和pd-1mrna中多个外显子-内含子基因座处的基因表达,随后耦合到生物信息程序,所述程序将整个基因中的基因表达归一化,以使得可极为准确地定量测量pd-1和pd-l1rna表达水平。此外,可对以下的升高rna水平进行量化:活化t细胞核因子1(nfatc1)、磷脂酰肌醇-4,5-二磷酸3-激酶催化亚基α(pik3ca)、磷脂酰肌醇-4,5-二磷酸3-激酶催化亚基δ(pik3cd)、pr域锌指蛋白1(prdm1)、磷酸酶与张力蛋白同源物(pten)、酪氨酸-蛋白磷酸酶非受体型11(ptpn11)、雷帕霉素机理靶(mtor)、缺氧诱导因子1-α(hif1a)和叉头框01类突变型(foxo1m)。此外,本发明方法可检测错配修复基因mlh1、pms2、msh6和mlh2的基因表达缺失。这些基因之一的功能缺失会引起基因组不稳定性,使得肿瘤表面新抗原的表达增加并由此增强对抗癌定向免疫疗法的响应速率。将视为与正常相比有所升高,并且因此指示阳性生物标记的任何特异性rna的水平示于表3a中。在另一实施例中,分析来自所述样品的dna,并检测编码生物标记的基因中的突变,所述突变对基因或基因产物的表达或功能产生影响。其中例如经由snv热点突变产生的突变的特定实例发现下表3b中所列的生物标记。确切地说,所述生物标记可在选自由以下组成的群组的基因中发现:间变性淋巴瘤激酶基因(alk)、b-raf基因(braf)、pd-l1基因(cd274)、表皮生长因子受体基因(egfr)、受体酪氨酸-蛋白激酶erbb-2或人类表皮生长因子受体2基因(erbb2)、成纤维细胞生长因子受体基因(fgfr)、kit原癌基因受体酪氨酸激酶基因(kit)、k-rasgtp酶基因(kras)、met原癌基因受体酪氨酸激酶(met)或n-ras原癌基因gtp酶基因(nras)。指示致癌突变的这些基因的特异性突变阐述在下表3b中。可使用如下文中所说明的常规技术检测这些突变。在一个特定实施例中,可检测由引起异常基因融合的基因重组产生的生物标记,并且这些生物标记列于下表3c中。例如,意识到alk基因可与一些癌症中的棘皮动物微管相关蛋白样4(eml4)基因的部分融合;fgfr基因可例如与其它癌症中的激酶形成融合物;并且met基因可变得与包括例如tfg、clip2或ptrz1的其它基因融合。任何所述融合基因/蛋白质的检测将提供阳性生物标记指示。在另一实施例中,所述分析鉴别出存在可能引起表达增加的拷贝数变异体。确切地说,分析来自所述样品的dna,并检测存在编码生物标记的基因的拷贝数变异。在这种情况下,适合的生物标记选自由以下组成的群组:erbb2、fgfr、fgfr1、fcfr2、fgfr3、fgfr4、cd273(pd-l2基因)或cd273,如表3d中所列。在此类情况下,注意到拷贝数增加,其可引起扩增或表达增加。在此类情况下,拷贝数增加得越多,所指示的敏感性越高。在这些情况下,正常拷贝数概述在表3d中。因此,与这些数字的偏离将指示对靶向pd-1/pd-l1路径的药剂的敏感性。用于测定生物标记的各种分析方法和技术宜尽可能在高通量测定平台中进行。在一个特定实施例中,将指示敏感性程度的算法应用于如上文所述获得的结果。确切地说,评分‘0’适用于显示相对于野生型或各种生物标记的正常表达谱无变化或变化极小的结果(例如在rna表达的情况下,每百万读段有0-500个归一化读段(nrpm)),而评分1适用于所注意到的任何突变或变异。在检测到特定基因的多个拷贝的情况下,可取决于所检测到的拷贝数分配较高评分,并且较高评分还可适用于极高rna表达水平变化的情况(例如>1500nrpm)。上文所论述的tmb评分可包括于所述算法中。在这种情况下,评分1可分配给<6mut/mb的‘低’tmb,评分2可分配给6-19mut/mb的中等tmb,并且评分3可分配给>20mut/mb的高tmb。所述算法的具体实例示于图1中。在这种情况下,评分1-2指示对靶向pd-1/pd-l1路径的药剂的极小敏感性;评分3-5指示对靶向pd-1/pd-l1路径的药剂的中度敏感性;并且超过6的评分指示对靶向pd-1/pd-l1路径的药剂的重大敏感性。适合地,将所述算法整合于用于推导生物标记数据的高通量系统中,以使得产生所述结果。所述系统将包含处理器和存储指令的存储器,以接收使用本发明方法获得的数据,使用上文所述的算法对其进行分析和转换以产生‘评分’,所述评分指示患者对使用靶向pd-1/pd-l1路径的组分的药剂的治疗的敏感性。这些结果可随后以适宜方式显示在图形界面上。在一些情况下,存储器将包含非暂时性计算机可读介质。所述系统和介质形成本发明的另一方面。使用本发明方法检测到的基因变异体可经由适合的生物信息平台与从临床试验中的药剂到经fda/ema批准的疗法的宽范围的靶向pd-1/pd-l1路径的潜在药剂相联系。通过这种方式鉴别后,肿瘤对所述抑制剂敏感的患者可因此使用适合药剂进行治疗。因此在另一方面,本发明提供一种用于治疗患有增生疾病的患者的方法,所述方法包含使用来自所述患者的肿瘤样品进行如上文所述的方法;基于生物标记的分析产生对患者的治疗或持续治疗的定制建议;并向所述患者施用适合疗法或治疗。确切地说,鉴别为对使用靶向pd-1/pd-l1路径的组分的药剂的治疗敏感或极为敏感的那些患者可用所述药剂治疗,而使用所述算法鉴别为几乎没有敏感性的那些患者将用替代药剂类型进行治疗。这些药剂将与正常临床操作同步施用。因此,在一个特定实施例中,本发明方法进一步包含基于所获得的结果产生针对治疗的定制建议。源自所述突变的整合信息实现待使用例如上文所述的系统和介质制备的疗法的定制建议,并且在此类情况下,这些还可显示在图形界面上。本发明方法解决了现有的针对抗pd-1/pd-l1/pd-l2定向免疫疗法的基于ihc的伴随诊断的问题和严重局限性。确切地说,所述方法适用于全自动化。特别是在使用上文所述的算法时,其可为定量的,并且不需要病理学家进行人类主观解释。在特定实施例中,本发明方法不需要病理学家输入pd-l1表达的人工评定结果。整个测试为全自动化的并且因此不会经历观察者间或观察者内变化。在一个特定实施例中,本发明方法提供与对抗pd-1/pd-l1/pd-l2免疫疗法的响应相联系的所有生物标记的综合和整合读出结果。如上文所述的算法可用于将所有这些预测性生物标记整合成多基因预测性评分(pps)(图1)。如上文所论述,pps评分0指示对抗pd-1/pd-l1/pd-l2免疫疗法无响应。评分1-2指示极小响应;评分3-5指示中度响应并且评分6-8指示较大响应。所述测定的这一定量性质意味着可鉴别精确切点来预测对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应。通过提供明晰的定量结果,避免进行基于ihc的测定的任何主观性和观察者间变化相关的解释。本发明方法能够在整合方法中评定整个pd-1/pd-l1信号传导轴,其无法使用单一ihc生物标记实现。使用本发明方法,可评定pd-1/pd-l1信号传导轴中的多种元件的活化/增加的表达,所述元件包括pd-1、pd-l1、pd-l2、nfatc1、pik3ca、pik3cd、prdm1、pten、ptpn11、mtor、hif1a、ifn-γ和foxo1。关键地是,本发明方法可以包括评定与对抗pd-1/pd-l1/pd-l2定向免疫疗法相联系的致癌突变。针对与抗pd-1/pd-l1定向免疫疗法相联系的基因异常、突变、融合和扩增评定宽范围的致癌基因(表3和图1a)。这些基因有多种为生长信号传导网络的组分。这些生长信号传导网络的致癌活化引起诱导pd-1配体,pd-l1和pd-l2。因此本发明方法可提供全自动测试,其已被设计成分析参与pd-1/pd-l1免疫调节性抗癌响应的所有组分,包括肿瘤细胞、免疫t细胞和抗原呈递细胞(aps)。这提供了关于所有细胞类型(肿瘤细胞和免疫细胞)和处于pd-1/pd-l1信号传导轴的所有水平下的参与pd-1/pd-l1/pd-l2免疫调节性癌症响应的所有组分的定量整合图。除了pd-1/pd-l1/pd-l2信号传导轴的综合分析之外,本发明方法已被设计成检测对于如上文所述的对抗pd-1/pd-l1定向免疫疗法的响应产生影响的所有致癌突变。具有致癌预测因子的源自pd-1/pd-l1/pd-l2信号传导轴的整合信息提供对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的最强有效预测因子。例如(i)突变和(ii)扩增pd-l1的检测还与(iii)pd-l1的表达增加相联系。这提供了对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的三个独立的但相联系的预测因子。通过将来自pd-1/pd-l1/pd-l2信号传导轴的信息与生长信号传导网络的致癌活化进行整合,本文所述的算法能够精确地鉴别出最有可能对抗pd-1/pd-l1/pd-l2定向免疫疗法作出响应的那些患者(表3a-d和图1)。本发明方法可使用高通量分析平台以患者肿瘤与来自经fda/ema批准的、esmo/nccn准则参考文献和全世界临床试验的所有阶段中的特异性靶疗法相匹配。申请者已利用赛默飞世尔(thermofisher)iontorrent平台来开发本发明测定。出于上文所解释的原因,目标在于通过分析如上文概述的这一路径的多种组分提供pd-1/pd-l1信号传导轴的综合图。使用本文所述的单个集成测试,并使用跨越两个免疫调节基因外显子/内含子边界的一组引物在两个基因的多个位点上进行设计,使它们能够扩增从常规福尔马林固定石蜡包埋的临床肿瘤物质/活检/切除标本中提取的降解rna物质。pd-1/pd-l1/pd-l2信号传导轴的这些多个组分的准确和精确测量使得可提供这一免疫调节性路径的定量整合图谱。来自这一测定平台的输出可随后用于提供预测对抗pd-1/pd-l1/pd-l2定向疗法的治疗性响应的这些免疫调节性生物标记的精确截止值。扩增子也已针对序列覆盖冗余被设计成重叠的以优化由常规诊断pwet样品获得的片段化dna模板的扩增。dna分析被设计成检测致癌突变和基因拷贝异常,其已鉴别为对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的预测因子。除了pd-1/pd-l1/pd-l2信号传导分子的rna表达分析之外,如果进行rna表达分析来检测致癌融合转录物,那么其鉴别为对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应的预测因子。适用于进行本发明方法的试剂盒具有新颖性并形成本发明的另一方面。这些试剂盒可包含检测表3中所列的3种或更多种生物标记所需的扩增引物的组合。此外,被安排成进行上文所述的方法的设备也具有新颖性并形成本发明的另一方面。确切地说,设备将包含用于进行如上文所述的dna及/或rna分析的构件,其连接到经编程以实施如上文所述的算法的计算机。以此方式编程的计算机或机器可读盒又形成本发明的另一方面,允许进行本文所述的方法的系统和非暂时性计算机可读介质同样如此。附图说明本发明现将通过实例参照伴随图解性附图进行具体描述,其中:图1为展示体现本发明的方法的示意图,所述方法包括用于解释结果并提供患者敏感性的指标的算法。具体实施方式本发明现将通过实例参照伴随图解性附图进行具体描述,其中:图1为展示体现本发明的方法的示意图,所述方法包括用于解释结果并提供患者敏感性的指标的算法。实例1材料和方法引物设计的综述:根据常规操作设计用于检测表3a-3d中所列的生物标记中的每一种的引物。一般来说,长度为18-30个核苷酸的引物为最佳的,其熔融温度(tm)在65℃-75℃之间。引物的gc含量应在40-60%之间,伴随引物的3'在c或g处结尾以促进结合。通过确保富gc域和富at域的平衡分布而使引物本身中的二级结构的形成降到最低。出于最佳引物性能,应避免引物内/引物间同源性。用于拷贝数检测的引物:引物被设计成跨越表3d中所列的基因中的精确区域,伴随每个基因有若干扩增子。针对这些扩增子中的每一个测量覆盖深度。拷贝数扩增和缺失算法是基于隐马尔可夫模型(hiddenmarkovmodel;hmm)。在进行拷贝数测定之前,针对gc偏好校正读段覆盖度并与预先配置的基线相比较。用于热点检测的引物:引物被设计成靶向易于发生如表3b中所列的致癌体细胞突变的特异性区域,并考虑上文所论述的一般位置。用于rna表达分析的引物:经由rt-pcr处理所提取的rna以产生互补dna(cdna),其随后使用特异性引物进行扩增。多个引物组被设计成跨越如表3a所列的经历表达分析的所有基因中的外显子/内含子界限。用于rna融合检测的引物:针对表3c中所列的每种融合设计一对经靶向的外显子-外显子断点测定引物。侧接融合体断点的引物产生特异性融合扩增子,其与参考序列进行比对以允许鉴别融合基因。表达不平衡测定实现在正常样品中监测等效表达水平,其中5'与3'测定之间的不平衡指示样品具有融合体断点。已开发出算法来整合源自常规石蜡包埋的组织活检体试样的这一经靶向的基因组分析的信息,以预测对抗pd-1/pd-l1/pd-l2定向免疫疗法的响应,如图1所示。dna和rna提取使用recoveraiitmambion提取试剂盒(目录号a26069)从切割呈10μm的ffpe卷曲或呈5μm的未染色载玻片上的切片中提取dna和rna。通过使1ml二甲苯与样品混合来进行两次二甲苯洗涤。对样品进行离心并去除二甲苯。在这之后用1ml纯乙醇进行2次洗涤。风干样品之后,将25μl消化缓冲液、75μl无核酸酶水和4μl蛋白酶添加到每个样品中。随后在55℃下消化样品持续3小时,之后在90℃下消化1小时。使120μl分离添加剂与每个样品混合,并将样品添加到收集管中的过滤筒中并进行离心。将过滤器移动到新收集管并保存在冰箱中以供在后期进行dna提取。保存流过物以供rna提取,并添加275μl纯乙醇,并将样品移动到收集管中的新过滤器并进行离心。用700μl第1洗液缓冲液洗涤之后,用dna酶如下处理rna;每个样品使用6μl10×dna酶缓冲液、50μl无核酸酶水和4μldna酶制备dna酶主混合物。将这添加到每个过滤器的中心并在室温下培育30分钟。培育之后,使用第1洗液,随后第2洗液/第3洗液进行3次洗涤,每次离心之后从收集管中去除洗涤缓冲液。将过滤器移动到新收集管并将洗脱液(加热到95℃)添加到每个过滤器中并培育1分钟。离心样品之后,丢弃过滤器并将流通中所收集的rna移动到新的低结合管。用第1洗液缓冲液洗涤过滤器中的dna,进行离心并丢弃流过物。用rna酶(50μl核酸酶水和10μlrna酶)处理dna并在室温下培育30分钟。如上伴随rna所述,完成三次洗涤并在95℃下加热的洗脱液中洗脱样品。dna和rna测量使用3.0荧光计以及rna高敏感性测定试剂盒(cat:q32855)和dsdna高敏感性测定试剂盒(cat:q32854)测量来自所提取的样品的dna和rna的量。在qubit测定管中使用与199μl合并的hs缓冲液和试剂合并的1μlrna/dna进行测量。将10μl标准品1或2与190μl缓冲液和试剂溶液合并以供对照。文库制备视需要将rna样品稀释到5ng/μl,并使用superscriptvilocdna合成试剂盒(cat11754250)逆转录到96孔盘中的cdna。针对所有样品制得2μlvilo、1μl10×superscriptiii酶混合物和5μl无核酸酶水的主混合物。96孔盘的每孔中使用8μl主混合物以及2μlrna。运作以下程序:温度时间42℃30分钟85℃5分钟10℃保持随后在每个样品孔中使用4μl的覆盖基因中的多个外显子-内含子基因座的6个rna引物、4μlampliseqhi-fi*1和2μl无核酸酶水进行cdna的扩增。使用以下程序将培养盘在热循环仪上运作30次循环:将dna样品稀释到5ng/μl并添加到ampliseqhi-fi*1、无核酸酶水中,并使用两个dna引物池(5μl池1和5μl池2)安设在96孔盘中。在热循环仪上运作以下程序:扩增之后,使用2μllibfupa*1部分消化扩增子,充分混合并放置在热循环仪上启用以下程序:温度时间50℃10分钟55℃10分钟60℃20分钟10℃保持(持续多达1小时)将4μl交换溶液*1、2μl稀释的ionxpress条码1-16(cat:4471250)和2μllibdna连接酶*1添加到每个样品中,其间添加每个组分时进行充分混合。在热循环仪上运作以下程序:温度时间22℃30分钟72℃10分钟10℃保持(持续多达1小时)随后使用30μl艾金科特(agencourt)ampurexp(拜奥麦克库尔特(biomeckcoulter)目录号(cat):a63881)纯化文库并培育5分钟。使用盘磁体,使用70%乙醇进行2次洗涤。随后在50μlte中洗脱样品。qpcr使用离子文库泰克曼(ionlibrarytaqman)定量试剂盒(cat:4468802)测量文库的量。使用大肠杆菌dh10bion对照文库的四个10倍连续稀释液(6.8pmol、0.68pmol、0.068pmol和0.0068pmol)产生标准曲线。以1/2000稀释每个样品,并一式两份地测试每个样品、标准品和阴性对照。针对每个样品在96孔快速热循环盘的孔中合并10μl2×taqman主混合物和1μl20×taqman测定物。将9μl的经1/2000稀释的样品、标准品或无核酸酶水(阴性对照)添加到盘中,并使用以下程序在abisteponeplustm机器(cat:4376600)上运作qpcr:使用te将样品稀释到100pmol,并将10μl每个样品收集到dna管或rna管。为了合并dna和rna样品,使用80:20dna:rna的比率。模板制备使用ions5ot2溶液和供应物*2初始化iononetouchtm2,并将150μl破坏溶液*2添加到每个回收管中。在无核酸酶水中进一步稀释所收集的rna样品(8μl所收集样品以及92μl水),并使用ions5试剂混合物*2以及无核酸酶水、ions5酶混合物*2、ionsphere粒子(isp)*2和稀释文库制得扩增主混合物。将主混合物连同反应油*2一起装入调适器中。所述仪器装载有扩增盘、回收管、路由器和装载有样品和扩增主混合物的扩增调适器。富集对于富集过程,使用280μltween*2和40μl1m氢氧化钠制得熔耗物(meltoff)。使用磁体用onetouch洗涤溶液*2洗涤myonetm链霉亲和素cl(cat:65001)。将珠粒悬浮于130μlmyone珠粒捕获溶液*2中。通过去除上清液,转移到新的低结合管来回收isp并且之后在800μl无核酸酶水中进行洗涤。离心样品并去除水的上清液之后,剩余20μl模板阳性isp。添加80μlisp再悬浮溶液*2,使最终体积为100μl。伴随以下将新的吸头(0.2ml管和8孔条)装载于onetouchtmes机器上:孔1:l00μl模板阳性isp孔2:130μl经洗涤的myonetm链霉亲和素c1珠粒,其再悬浮于myone珠粒捕获溶液中孔3:300μliononetouches洗涤溶液*2孔4:300μliononetouches洗涤溶液孔5:300μliononetouches洗涤溶液孔6:空孔7:300μl熔耗物孔8:空在耗时大约35分钟进行运作之后,对富集的isp进行离心,去除上清液并用200μl无核酸酶水洗涤。另一离心步骤并去除上清液之后,剩余l0μlisp。添加90μl无核酸酶水并使珠粒再悬浮。测序使用ions5试剂筒、ions5清洗溶液和ions5洗涤溶液*2初始化ions5systemtm(cat:a27212)。将5μl对照isp*2添加到富集样品中并充分混合。对管进行离心并去除上清液,留下样品和对照isp。将15μlions5退火缓冲液*2和20μl测序引物*2添加到样品中。针对在95℃下退火2分钟并在37℃下退火2分钟的引物,将样品装载于热循环仪上。热循环之后,添加l0μlions5装载缓冲液*2并混合样品。使用500μlions5退火缓冲液*2和500μl无核酸酶水*2制得50%退火缓冲液。随后将整个样品装入ion540tm芯片(cat:a27766)的装载口中,并在芯片离心机中离心10分钟。在这之后,将l00μl泡沫体(使用49μl50%退火缓冲液和1μl发泡溶液*2制得)注入所述口中,之后将55μl50%退火缓冲液注入芯片孔中,从出口孔中去除过量液体。将芯片离心30秒,伴随芯片凹口朝外。重复这一发泡步骤。在装载口中使用100μl冲洗溶液(使用250μl异丙醇和250μlions5退火缓冲液制得)冲洗芯片两次,并从出口孔中去除过量液体。随后在装载口中用50%退火缓冲液进行3次冲洗。将60μl50%退火缓冲液与6μlions5测序聚合酶*2合并。随后将65μl聚合酶混合物装入所述口中,培育5分钟并装载于s5仪器上以进行测序,其耗时大约3小时并耗时16小时进行数据传送。*1来自ionampliseqtm文库2.0(cat:4480441)*2来自ion540tmot2试剂盒(cat:a27753)数据分析dnacnv分析:拷贝数变异(cnv)表示其中基因组的区段已被重复(增加)或缺失(丢失)的一类变异。较大的基因组拷贝数不平衡可以在子染色体区域到整个染色体范围内。在ions5系统上处理原始数据并将其转移到torrent服务器,以使用昂科曼综合分析基准(oncominecomprehensiveassaybaseline)v2.0进行初步数据分析。这一插件包括于torrentsuite软件中,其伴随有每个iontorrenttm测序器。使用基于隐马尔可夫模型(hmm)的算法进行拷贝数扩增和缺失检测。算法使用横跨基因组的读段覆盖度来预测拷贝数。在进行拷贝数测定之前,针对gc偏好校正读段覆盖度并与预先配置的基线相比较。报告每个样品的所有成对差的绝对值的中值(mapd)评分并用于评定样品变化并界定数据是否适用于拷贝数分析。mapd为拷贝数变异的每个测序运作估计值,类似于标准差(sd)。如果假定log2比率相对参考物,恒定sd以平均值0正态分布,那么mapd/0.67等于sd。然而,不同于sd,针对藉由已知病状,如癌症诱发的log2比率的高生物变化性,使用mapd为稳健的。具有高于0.5的mapd评分的样品在验证cnv读取之前应谨慎地检查。归一化之后拷贝数分析产生的结果可以从原始数据中可视。体细胞cnv检测提供每个拷贝数区段的置信界限。所述置信度为拷贝数小于所界定的给定拷贝数的估算百分比概率。针对每个cnv给定较低和较高百分比和所界定的相应的拷贝数值。还陈述每个cnv的置信区间,并且归类为存在归一化之后5%置信度值≥4的拷贝数>6的扩增和95%ci≤1的缺失。dna热点分析:在ions5系统上处理原始数据并将其转移到torrent服务器,以使用定制工作流程进行初步数据分析。进行原始数据与参考基因组的映射和比对并随后根据bed档注释热点变异体。覆盖度统计数据和其它相关qc标准界定在vcf档中,其包括使用大量的公开来源组的注释。过滤参数可适用于鉴别通过qc阈值的那些变异体,并且这些变异体可以在igv上可视。一般来说,以>10%替代等位基因读段对变异体进行分类的规则,并且呈>10独特读段的归类为‘被检测到’。若干计算机模拟工具用以评定所鉴别的变异体的致病性,这些包括phylop、sift、grantham、cosmic和polyphen-2。rna表达分析:在ions5系统上处理原始数据并将其转移到torrent服务器,以使用ampliseqrna插件进行初步数据分析。这一插件包括于torrentsuite软件中,其伴随有每个iontorrenttm测序器。ampliseqrna插件使用torrent映射比对程序(tmap)。针对iontorrenttm测序数据优化tmap以供相对于含有ampliseq试剂盒所靶向的所有转录物的定制参考序列组比对原始测序读段。测定特定信息包含于订制bed档中。为了维持特定性和敏感性,tmap实施两步映射方法。首先,采用四个比对算法,bwa-短、bwa-长、ssaha和超最大精确匹配来鉴别一系列候选映射位置(cml)。使用史密斯沃特曼算法(smithwatermanalgorithm)进行另一比对过程以发现最终最佳映射。作为ampliseqrna插件的一部分,使用samtool(samtoolsview-c-f4-lbed_filebam_file)进行所靶向基因的原始读段计数。通过插件将给定样品的ionampliseqrna归一化结果自动计算为每个基因每百万映射读段或rpm的映射读段数。这一数字随后经log2转变成每百万的归一化读段(nrpm)。订制bed档经格式化而含有映射参考物中的每个转录物的每个扩增子的核苷酸位置。与预期扩增子位置比对并符合如最小比对长度的过滤标准的读段报告为“有效”读段百分比。“所检测到的目标”被定义为呈总目标数的一定百分比的所检测到的扩增子数(≥10读段计数)。映射、比对和归一化之后,ampliseqrna插件提供关于qc度量值的数据、可视化曲线和每个基因的归一化计数,其对应于包括详述制表符定界文本文档中每个基因的读段计数的可下载文档的链接的基因表达信息。与给定基因目标比对的读段数表示称作“计数”的表达值。这一另外插件分析包括具有至少1、10、100、1,000和10,000个计数的基因(扩增子)数的每个条码的输出以实现测定每个样品的动态范围和敏感性。上文信息的汇总表,包括总映射读段的每个条码的映射统计数据、关于目标的百分比和所检测到的小组基因(“所检测到的目标”)的百分比可在torrentsuite软件中查看以快速评估运作和文库性能。界定pd-1和pd-1表达的截止点:使用一系列呈现每个组织器官系统(例如脑、肺脏、乳房、结肠、卵巢、前列腺膀胱)的正常组织样品测定pd-1和pd-l1rna表达值,并使用免疫聚焦(immunofocus)测试与pd-1和pd-l1蛋白质表达水平相关联,所述测试利用使用兔单克隆抗体(e1l3,cell信号传导)的免疫组织化学测定。这些数据提供在0-500nrpm范围内的正常组织的基线pd-1和pd-l1rna表达水平。随后针对一系列肿瘤类型测定pd-1和pd-l1rna表达并将其与pd-1和pd-l1免疫聚焦(immunofocus)蛋白质表达水平相关联。如正常组织中所观察到的显示不存在或低pd-1/pd-l1rna表达水平(即,0-500nrpm)的肿瘤与如由免疫聚焦(immunofocus)测定的<1%的pd-1/pd-l1肿瘤比例评分相关。显示如由免疫聚焦(immunofocus)评定的>50%的高肿瘤比例评分的肿瘤与在1500-2000nrpm范围内的高rna表达相关。对于在1%与50%之间的肿瘤比例评分,观测到pd-1和pd-l1rna表达的相应增加量在500-1500nrpm范围内。这些数据最终显示,可以通过定量测定测量pd-1和pd-l1rna表达的临床上相关水平,并且所述水平因此能够替代当前用作抗pd-1和抗pd-l1定向疗法的伴随诊断的半定量和主观的基于ihc的测定。这提供准确鉴别可能受益于免疫治疗剂的那些患者的强有效和创新的新方法。表1ihc=免疫组织化学;pd-1=程序性细胞死亡蛋白;pdl-1=程序性死亡配体1;nsclc=非小细胞肺癌表2pd-l1ihc22c3pharmdx结果的诊断状态表3a基于异常rna表达的生物标记表3bdna突变表3c基因融合表3ddna拷贝数变异体当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1