用于制造低粘度纤维素醚的简化方法与流程
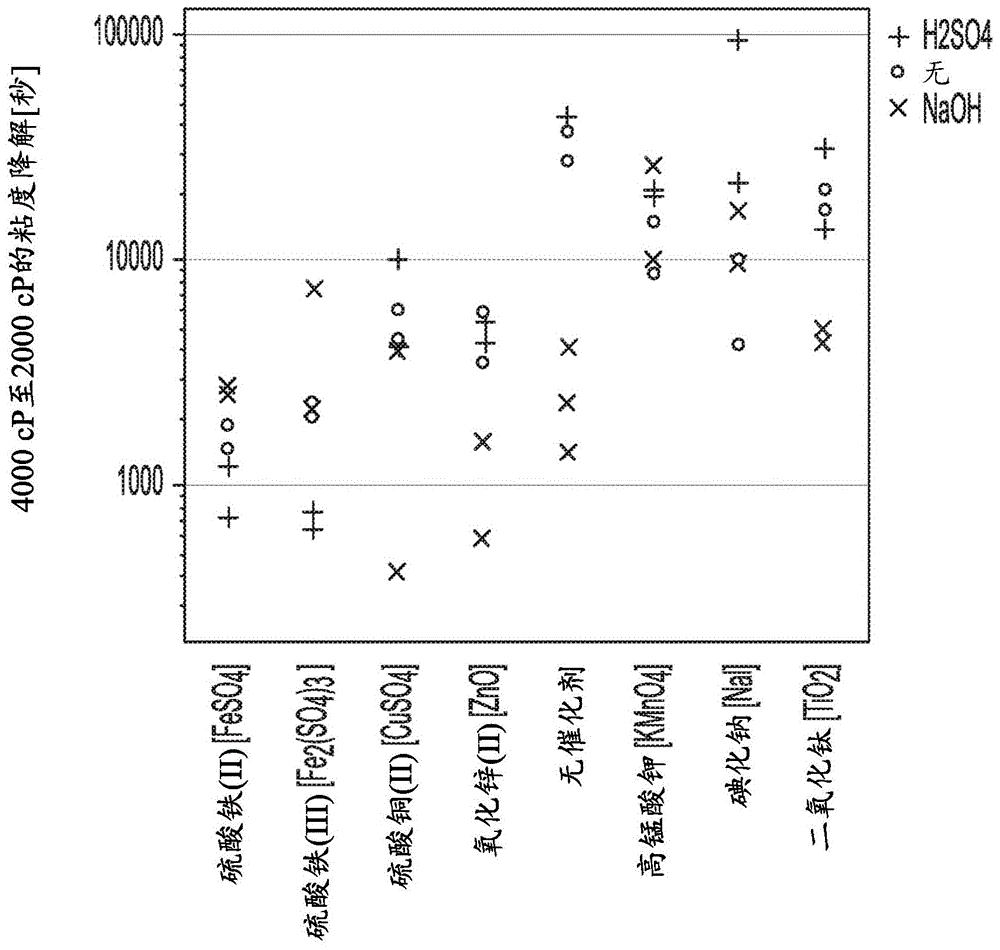
发明背景本发明涉及一种用于由较高粘度的纤维素醚制造低粘度纤维素醚的单一方法。引言两步法通常用于制备具有中等和低粘度的纤维素醚。第一步是制备初始纤维素醚并且然后洗涤、配混并干燥所述初始纤维素醚以形成初始纤维素醚粉末。第二步是使所述初始纤维素醚粉末经受酸水解、中和和干燥以将所述初始纤维素醚转化为较低粘度的纤维素醚。所述两步法需要两个反应步骤、两个干燥步骤和两组设备。结果,所述两步法是设备密集的并且需要能量来干燥纤维素醚产物两次。此外,所述两步法典型地需要使用卤代酸进行酸水解以降低纤维素醚的粘度。此种水解步骤不希望地需要处理腐蚀性酸并且倾向于产生以下纤维素醚产物,所述纤维素醚产物由于最终产物中的残留酸而在粘度上不稳定,或者需要大量的淬灭努力以从最终产物中清除残留酸。希望能够确定一种用于生产纤维素醚并且然后降低所述纤维素醚的粘度的方法,其全部是单一连续过程在最后具有单一干燥步骤以便减少设备需求和能量需求。此外,希望此种方法不包括使用卤代酸进行酸水解以便避免处理腐蚀性酸以及上述由于最终产物中残留酸的挑战。技术实现要素:本发明的方法提供了一种用于生产纤维素醚并且然后降低所述纤维素醚的粘度的方法,其全部是单一连续过程在最后具有单一干燥步骤。此外,本发明提供了可以不包括使用卤代酸进行酸水解的这样的方法。本发明的方法可以是连续的,包括合成较高粘度的纤维素醚、降低纤维素醚粘度和分离较低粘度的纤维素醚,在最后具有唯一的干燥步骤以分离较低粘度的纤维素醚。本发明是出人意料地和出乎意料地发现以下的结果:在洗涤纤维素醚产物之后并且不干燥纤维素醚产物的情况下可以引入氧化还原活性的过渡金属基催化剂和含过氧基的氧化剂以便将纤维素醚产物转化成较低粘度纤维素醚而无需干燥、分离或单独的酸水解步骤。所述方法可以不包括引入酸和淬灭碱来进行水解反应以降低纤维素醚粘度。此外,进一步出人意料地和出乎意料地发现:可以在本发明的方法中引入增强剂以便减少纤维素醚的变色以便提供更白的纤维素醚产物。增强剂是选自由以下组成的组的一种或多种组分:5-取代的3,4-二羟基呋喃酮(如抗坏血酸和异抗坏血酸)、偏亚硫酸氢盐、亚硫酸盐、硫代硫酸盐和二氧化硫。进一步出人意料地且出乎意料地发现:冲击研磨干燥最终纤维素醚产物特别有利于同时从最终纤维素醚中去除水分和残留氧化剂而不大量浓缩氧化剂和导致所得纤维素醚的不希望的降解。甚至更出人意料地和出乎意料的是,在冲击研磨之前将水添加到纤维素醚产物中实际上增加了在冲击研磨干燥步骤期间去除氧化剂的效率。甚至此外,出人意料地和出乎意料地发现:可以在本发明的方法中引入淬灭剂以便稳定所得的纤维素醚团使其在延长的储存时不降解。在第一方面,本发明是一种用于制备纤维素醚的方法,所述方法包括:(a)烷基化和醚化纤维素以形成初始纤维素醚;(b)洗涤和过滤所述初始纤维素醚以产生经洗涤的纤维素醚;(c)任选地,使所述经洗涤的纤维素醚粒化;(d)将所述经洗涤的纤维素醚配混以形成经配混的纤维素醚团;(e)任选地,进一步将额外的组分混入所述经配混的纤维素醚中;以及(f)干燥所述经配混的纤维素醚团以获得具有的粘度低于所述初始纤维素醚的最终纤维素醚;其中所述方法的特征在于:(i)在以下步骤中的至少一个期间引入水性催化剂,所述水性催化剂为氧化还原活性过渡金属基催化剂:粒化(c)、配混(d)、混合步骤(e)和干燥步骤(f);以及(ii)在以下步骤中的至少一个期间引入含过氧基的氧化剂:粒化(c)、配混(d)、混合步骤(e)和干燥步骤(f);以及(iii)在以下步骤中的至少一个期间引入水性增强剂:粒化(c)、配混(d)和混合步骤(e);其中所述水性增强剂选自由以下组成的组:5-取代的3,4-二羟基呋喃酮、偏亚硫酸氢盐、亚硫酸盐、硫代硫酸盐和二氧化硫;以及(iv)在步骤(a)中的所述烷基化之后并且在步骤(f)中所述干燥所述经配混的纤维素醚以获得所述最终纤维素醚之前,不包括干燥和分离纤维素醚。本发明的方法可用于有效地制造纤维素醚,尤其是具有8,000毫帕*秒(mpa*s)及更低的粘度的那些纤维素醚。附图说明图1、3、5、7和9提供了实例的各种溶液的降解半衰期(4000mpa*s至2000mpa*s所需的时间)的图。图2、4、6、8和10提供了实例的各种溶液的变色图。图11和12示出了如实例中所述的不同降解反应运行的对比粘度下降。图13示出了来自实例的阴性对照的随时间推移的粘度曲线。具体实施方式“和/或”意指“和、或可替代地”。除非另外说明,否则范围包括端点。除非日期与测试方法号一起指示为带连字符的两位数字,否则测试方法是指截止至此文件的优先权日的最近的测试方法。对测试方法的提及包含对测试学会和对测试方法号的提及。通过以下缩写中的一个来提及测试方法组织:astm是指astm国际协会(以前称为美国材料及试验学会(americansocietyfortestingandmaterials));en是指欧洲标准(europeannorm);din是指德国标准化学会(deutschesinstitutfürnormung);并且iso是指国际标准化组织(internationalorganizationforstandardization)。“纤维素醚”包括烷基纤维素醚和羟烷基纤维素醚。作为特定实例,纤维素醚包括以下任何一种、或一种以上的组合:甲基纤维素、乙基纤维素、羟乙基甲基纤维素、羟丙基甲基纤维素、羟乙基甲基纤维素、乙羟基乙基纤维素和羟丁基甲基纤维素。特别引人关注的是烷基纤维素醚,诸如甲基纤维素和羟丙基甲基纤维素。在本文中,除非在传授内容的上下文中另外指出,否则通过经由usp40-nf35第4552页在“hypromellose[羟丙甲纤维素]”下阐述的方法来制备2重量%(wt%)的纤维素醚水溶液来确定所述纤维素醚在20℃下的粘度。纤维素醚的粘度随纤维素醚的分子量变化,使得较高分子量的纤维素醚具有比较低分子量的纤维素醚高的粘度。较高分子量的纤维素醚的水解产生较低分子量/较低粘度的纤维素醚。在本文中,毫帕*秒(mpa*s)和厘泊(cp)的单位可以互换。本发明是一种用于通过制造初始纤维素醚并且然后降低所述初始纤维素醚的粘度来生产纤维素醚的方法。与当前的方法不同,本发明的方法不需要在初始纤维素醚制造后和降低其粘度之前分离出所述初始纤维素醚,也不需要使用卤代酸水解来降低纤维素醚粘度。实际上,本发明令人希望地不包括这些方法步骤中的任何一个。本发明的方法包括以下步骤:(a)烷基化和纤维素醚以产生经洗涤的纤维素醚;(c)任选地,使所述经洗涤的纤维素醚粒化;(d)将所述经洗涤的纤维素醚配混以形成经配混的纤维素醚团;(e)任选地,进一步将额外的组分混入所述经配混的纤维素醚中;以及(f)干燥所述经配混的湿纤维素醚团以获得具有的粘度低于所述初始纤维素醚的最终纤维素醚。方法步骤(c)和(e)是任选的,这意味着它们对于本发明的最宽范围不是必需的,但是任何一个或两个可以作为本发明的一部分被包括。步骤(a):纤维素醚的烷基化和醚化在本发明的最宽范围内,形成初始纤维素醚的纤维素醚的烷基化和醚化不受限制并且可以通过任何方法进行。例如,us6261218在第3栏第9行至第67行中公开了用于纤维素醚的烷基化和醚化的合适方法,其适合用于本发明中来制备初始纤维素醚。适用于纤维素醚的烷基化和醚化的一般方法如下:提供初始呈粉末形式或颗粒的纤维素浆,典型地是棉浆或木浆。在反应器中在碱性氢氧化物、优选氢氧化钠存在下烷基化所述纤维素浆。例如,烷基化可通过在含有水性氢氧化物的浴或搅拌槽中浸泡或将水性氢氧化物直接喷在干浆上而发生。水性氢氧化物优选以基于水的重量的30-70重量%的碱性氢氧化物含量使用。保持速率优选为5至90分钟。烷基化的温度优选为30摄氏度(℃)至60℃。通过混合和搅动,在浆中实现均匀的溶胀和碱分布。可以将烷基化反应器的顶部空间抽空或用惰性气体如氮气部分或基本上吹扫以控制纤维素醚产物的解聚。未反应的碱性氢氧化物可用酸如盐酸、硝酸或乙酸中和,或可用稍过量的醚化剂中和。适用于烷基化的纤维素醚的醚化的一般方法如下:将烷基化的纤维素醚放入反应器中(如果尚未放入反应器中),并将反应器中的压力升高至650千帕(kpa)或更高、更典型地690kpa或更高、700kpa或更高、750kpa或更高或甚至800kpa或更高的压力而同时至典型地4000kpa或更低、3500kpa或更低、3000kpa或更低、2500kpa或更低、或者甚至2100kpa或更低的压力持续约0.5至16小时。典型的醚化剂包括低级烷基卤化物和环氧化物,如氯甲烷、氯乙烷、环氧乙烷、环氧丙烷、环氧丁烷及其混合物。例如,可以使用氯甲烷来制造甲基纤维素,并且可以使用氯甲烷和环氧丙烷的混合物来制造羟丙基甲基纤维素。氯甲烷的使用导致形成氯化钠副产物。优选地,添加稍过量的醚化剂以与从烷基化剩余的任何未反应的碱性氢氧化物反应。所得的纤维素醚是初始纤维素醚并且令人希望地具有200毫帕*秒(mpa*s)或更高、优选地4000mpa*s或更高并且同时典型地400,000mpa*s或更低的粘度。令人希望地,初始纤维素醚具有如由式i表示的结构,其中纤维素醚具有如括号中所指定的重复单元:其中r1、r2和r3每次出现时独立地选自由以下组成的组:氢以及直链或支链的c1-c5烷基,所述烷基任选地被一个或多于一个c2-c5直链或支链的烷氧基或羟基取代,前提是重复单元r1、r2和r3中的至少一个各自不为氢。步骤(b):初始纤维素醚的洗涤和过滤洗涤初始纤维素醚以去除盐和烷基化/醚化的其他反应副产物。盐可溶于其中的任何溶剂均适用于洗涤,但是由于其可获得性和环境相容性,热水是优选的。令人希望地,洗涤醚化反应器和/或醚化反应器的下游。在洗涤之前或之后,可以通过暴露于蒸汽来汽提纤维素醚以减少残留的有机物含量。洗涤后过滤初始纤维素醚。令人希望地,通过本领域已知的任何方法进行过滤。例如,离心过滤法、压滤、真空过滤、加压滤板法都是从初始纤维素醚中过滤洗涤液的合适手段。步骤(c):经洗涤的纤维素醚的任选的粒化经洗涤的纤维素醚可以并且令人希望地在配混以形成经配混的纤维素醚团之前粒化。粒化用于将经洗涤的纤维素醚团聚成更大的微粒形式。粒化可以通过适用于使纤维素醚粒化的任何方法进行。例如,使用例如球磨机或冲击式粉碎机的研磨是合适的粒化方法。当使用球磨机或冲击式粉碎机时,典型的保留时间为约20分钟至约120分钟。典型地,如通过机械筛确定的,经洗涤的纤维素醚具有25至1000微米的平均粒度,其中平均粒径对应于其中一半质量保留在筛上并且一半质量通过筛的粒径。步骤(d):配混经洗涤的纤维素醚将所述经洗涤的纤维素醚配混以形成经配混的纤维素醚团。通常,为了使纤维素醚中的水分均化到纤维素醚中以形成团状材料,配混通过连续的高剪切混合发生。用于高剪切混合的合适手段包括配混挤出机,如双螺杆挤出机。其他合适的高剪切混合器包括捏合机和粒化机。在配混期间,可以向纤维素醚中添加(或不添加)额外的水以促进配混。相对于经洗涤的纤维素醚的总重量(纤维素醚和水分的重量),纤维素醚的水分含量典型地为20-90、30-75、40-75wt%的水。较低的温度对于配混是令人希望的以促进水吸收到纤维素醚中。通常,在2℃-80℃的温度下进行配混步骤(e):任选的通过缓冲罐进料经配混的纤维素醚可将经配混的纤维素醚从配混步骤进料到容器(“缓冲罐”)中以缓冲将经配混的纤维素醚进料到干燥步骤的速率。使用缓冲罐是令人希望的以对纤维素醚中的组分反应提供停留时间。使用缓冲罐还是令人希望的以抑制上游进料速率的变化,使得可以将经配混的纤维素醚以更恒定的速率进料到干燥步骤中。纤维素醚在缓冲罐中的停留时间令人希望地为1至15分钟。缓冲罐令人希望地包括低剪切搅动器或混合器以保持经配混的纤维素醚可移动。合适的缓冲罐的实例包括具有入口和出口以及桨式搅动器的罐,所述桨式搅动器使经配混的纤维素醚保持朝向缓冲罐的出口移动。步骤(f):干燥经配混的纤维素醚团干燥所述经配混的纤维素醚团以获得具有的粘度低于所述初始纤维素醚的最终纤维素醚。干燥有利地通过冲击研磨经配混的纤维素醚团来进行。冲击研磨干燥特别有利于同时去除水分和残留的氧化剂而不大量浓缩氧化剂(其在其他形式的干燥情况下可能发生)。去除氧化剂对于避免纤维素醚的不令人希望的降解是有价值的,所述降解可能导致最终纤维素醚的粘度在干燥过程期间漂移。在干燥期间有效去除氧化剂防止替代方法的不令人希望的方法特征,如:(i)由于大量加热以去除水分和氧化剂而使纤维素醚变色;(ii)由于使用洗涤步骤去除氧化剂而延长了干燥时间;以及(iii)由添加大量的淬灭剂以去除氧化剂而导致的纤维素醚产率降低。通过冲击研磨有效地去除氧化剂促进更大地控制最终纤维素醚的粘度和最终纤维素醚的粘度稳定性而对替代方法没有不利影响。可替代地,纤维素醚的干燥可以通过本领域已知的任何其他手段进行,如蒸汽管干燥、接触干燥和对流干燥(如闪蒸干燥)而不是冲击研磨。在通过此类方法干燥之前,将经配混的纤维素醚散布到糊中促进呈蒸汽管干燥、接触干燥和对流干燥过程形式的干燥过程。出人意料地,在步骤(f)中干燥之前,向纤维素醚中添加水,尤其是当通过冲击研磨进行干燥时。在干燥步骤之前添加水实际上提高了在干燥步骤期间去除氧化剂的效率。因此,希望在配混步骤(d)或任选的步骤(e)期间添加水。令人希望地,在步骤(d)和(e)期间添加的水的总量是这样的,即,使得基于水和纤维素醚组分的组合重量,干燥前的水含量为45-75wt%。方法特征本发明方法的特征至少在于以下四个特征:(i)在以下步骤中的任何一个或多于一个的任何组合期间添加(即,引入)水性催化剂:粒化(c)、配混(d)、混合步骤(e)和干燥步骤(f);(ii)在以下步骤中的任何一个或多于一个的任何组合期间添加(即,引入)含过氧基的氧化剂:粒化(c)、配混(d)、混合步骤(e)和干燥步骤(f);(iii)在以下步骤中的任何一个或多于一个的任何组合期间添加(即,引入)水性增强剂:粒化(c)、配混(d)和混合步骤(e);以及(iv)所述方法在步骤(a)中的所述烷基化之后并且在步骤(f)中所述干燥所述经配混的纤维素醚以获得所述最终纤维素醚之前,不包括干燥和分离纤维素醚。这些特征中的“引入”意指添加到指定步骤的纤维素醚组分中。“纤维素醚组分”包括初始纤维素醚、经洗涤的纤维素醚和经配混的纤维素醚团。方法特征(i):水性催化剂添加水性催化剂是在水中的氧化还原活性过渡金属基催化剂。令人希望地,催化剂是选自由硫酸铁(ii)和硫酸铁(iii)组成的组的任何一种或多于一种的任何组合。优选地,铜盐是一种或多种硫酸铜。令人希望地,引入水性催化剂以建立0.01重量%(wt%)或更高、0.0.05wt%或更高、0.1wt%或更高或0.5wt%或更高并且同时通常为1wt%或更低的总催化剂浓度(即,过程中引入的所有催化剂的总和),其中wt%催化剂是相对于干纤维素醚组分重量。方法特征(ii):氧化剂添加所述含过氧基的氧化剂令人希望地是选自过氧化氢、无机过硫酸盐和有机过硫酸盐的一种或多于一种的任何组合。令人希望地,将含过氧基的氧化剂以以下总浓度(过程中引入的所有含过氧基的氧化剂的总和)引入到过程中:引入过程中的总催化剂重量的一倍或多倍、优选5倍或更多倍、甚至6倍或更多倍并且同时典型地引入过程中的总催化剂重量的500倍或更小、更典型地100倍或更小、甚至更典型地50倍或更小并且可以是30倍或更小、25倍或更小并且甚至20倍或更小。方法特征(iii):水性增强剂添加水性增强剂是在水中的一种或多于一种芬顿(fenton)增强剂的组合。芬顿增强剂是选自由以下组成的组的任何一种或多于一种组分:5-取代的3,4-二羟基呋喃酮、偏亚硫酸氢盐、亚硫酸盐、硫代硫酸盐、抗坏血酸盐和二氧化硫。合适的5-取代的3,4-二羟基呋喃酮的实例包括抗坏血酸和异抗坏血酸及其异构体。在过程期间引入的水性增强剂的总量令人希望地足以实现以下总芬顿增强剂浓度(即,过程期间引入的所有芬顿增强剂的量):过程期间引入的总催化剂的重量的0.01或更高、优选地0.05或更高、更优选地0.08或更高倍并且可以是0.10或更高、1或更高、5或更高、10或更高、25或更高、50或更高并且甚至75或更高倍并且同时通常为过程期间引入的总催化剂的重量的100或更小、75或更小、50或更小、25或更小、10或更小并且可以是5或更小并且甚至1或更小倍。相对于没有增强剂的类似方法,所述增强剂对本发明的方法至少提供以下益处:(1)最终纤维素醚(即,白色最终纤维素醚)的更快降解。更快降解是令人希望的以使反应更有效且成本更低。对于生产用于其中白度是重要的应用(如药物应用)和其中使用后续着色并且无论纤维素醚批次如何都需要准确实现可重复的颜色的应用的纤维素醚,较少的变色也是有价值的。方法特征(iv):在步骤(f)之前不包括干燥和分离纤维素醚本发明的方法有利地可以是连续方法,其从烷基化和醚化以形成纤维素醚到降低纤维素醚的粘度一路上带着纤维素浆而无需沿途干燥或分离纤维素醚。这意味着本发明的方法避免了目前用于制备纤维素醚并且然后降低其粘度的方法中所需的干燥和分离步骤。实际上,本发明的方法可以是一种连续方法,其从烷基化和醚化纤维素浆以形成初始纤维素醚开始到降低初始纤维素醚的粘度和分离降低粘度的纤维素醚。在那方面,本发明的方法在步骤(a)之后和步骤(f)之前的任何地方都不包括干燥和分离纤维素醚。此外,步骤(a)中形成的纤维素醚可以经过本发明的方法而没有任何水含量的降低直到在步骤(f)中干燥。这样,所述方法消除了对用于烷基化/醚化和降解(粘度降低)的单独反应器的需求。通过消除中间的干燥和分离步骤,此种单一方法提高了生产中至低粘度纤维素醚的能量和时间效率。任选的淬灭剂所述方法可以进一步包括在配混期间在配混步骤(d)期间或之后的任何时间在添加催化剂、氧化剂和增强剂后添加淬灭剂。通过消耗残留的氧化剂和/或催化剂,猝灭剂的添加对最终纤维素醚粘度提供了进一步的稳定性。任选的淬灭剂可以是选自下述四组淬灭剂的任何一种或多于一种组分的任何组合。每组淬灭剂通过同一组内或者来自另一组的一种或多于一种淬灭剂起作用,或者可以使用选自所述组之一的仅一种单一淬灭剂。淬灭剂组i:偏亚硫酸氢盐、亚硫酸盐、硫代硫酸盐和二氧化硫。来自组i的猝灭剂表现得很像增强剂添加剂并且增强反应速率以消耗氧化剂。当使用来自淬灭剂组i的淬灭剂时,淬灭剂浓度相对于在过程期间引入的氧化剂典型地为1:1和0.001:1的摩尔比。当淬灭剂与所列的增强剂相同时,将材料用作“淬灭剂”是明显的,因为在添加增强剂并引入氧化剂和催化剂后的一段时间将其引入。淬灭剂组ii:ec1.11.1类过氧化物酶(如由国际生物化学和分子生物学联合会命名委员会(nomenclaturecommitteeoftheinternationalunionofbiochemistryandmolecularbiology)定义),如过氧化氢酶,以及锰(ii)至锰(vii)的盐氧化物和二氧化物。来自此组ii的猝灭剂通过不引入羟基自由基中间体的机理催化过氧化氢降解为水和氧气。因此,来自组ii的淬灭剂可用于去除过氧化氢氧化剂以终止纤维素醚的粘度降低。当使用时,来自淬灭剂组ii的淬灭剂典型地以相对于在过程期间引入的氧化剂0.01:1至0.0001:1的摩尔比的浓度存在。淬灭剂组iii:一种或多于一种螯合剂(如乙二胺四乙酸(edta))的任何组合,相对于催化剂的摩尔比为4:1和1:4,和/或柠檬酸,相对于催化剂的摩尔比为4:1至1:4,或0.05至0.2毫摩尔/克纤维素醚。螯合剂通过与金属催化剂络合而用作淬灭剂,从而减慢或停止纤维素醚的降解反应。淬灭剂组iv:抗坏血酸和异抗坏血酸中的一种或两种。组iv的淬灭剂用于加速过氧化氢的消耗,从而以不有助于纤维素醚的粘度降低的方式消耗氧化剂。当作为淬灭剂存在时,淬灭剂组iv材料典型地处于0.05至0.2毫摩尔/克纤维素醚的摩尔浓度。本发明方法有利地含有淬灭剂以产生多于一种提及的淬灭剂的稳定的最终组合。例如,本发明的方法可以包括添加edta或不添加edta。此外,所述方法可以不包括所有提及的淬灭剂。本发明的方法可以不包括钴和锰的盐。本发明的方法可以不包括除了铁、铜和锌的那些盐以外的任何过渡金属盐。实例为了方便实验,以下实例用初始纤维素醚粉末开始进行,所述初始纤维素醚粉末被水合以代表并模拟所要求保护的本发明的经洗涤的初始纤维素醚。预期以下实例的结果完全代表通过如上所述的将纤维素烷基化和醚化以形成初始纤维素醚以及如上所述的洗涤并过滤所述纤维素醚并且然后经由粒化步骤或直接行进到如下文所述降解步骤而获得的结果。换句话说,以下结果与初始纤维素醚是否在降解之前被干燥和分离,或者是否将初始纤维素醚从烷基化/醚化后的洗涤和过滤转移到降解(直接或经由粒化步骤)无关。compexa-h和ex1:催化剂+增强剂+氧化剂的协同作用对于对比实例(compex)a-h和实例(ex)1中的每一个,通过以timothythomson等人在us4845206中描述的方式水合具有2663-4970mpa*s的粘度、28-30的甲氧基wt%和7-12的羟丙基wt%的羟丙基甲基纤维素(例如,methoceltme4m级羟丙基甲基纤维素;methocel是陶氏化学公司(thedowchemicalcompany)的商标)制备200克处于50wt%水分水平的纤维素醚湿饼。在25℃下将纤维素醚湿饼装载到5升lodigeploughshare混合器中并且打开混合器。将指定量的硫酸铁(iii)和/或抗坏血酸(参见表1)溶解在20克去离子水中,并在混合的同时以60毫升/分钟的速率将溶液添加到纤维素醚湿饼中。在25℃下继续混合25分钟以形成。将团混合物转移到linden双z批次实验室捏合机中,并在25℃下在混合的同时以小于1分钟添加指定量的30%过氧化氢(参见表1)。将所得团在25℃下混合30分钟。对于含有过氧化氢的样品,使用商业过氧化氢酶消耗残留的过氧化物以实现通过淀粉-碘化物测试条的不含过氧化物的团。从捏合机中移出团,手动破碎成小块,并在协定烘箱中在75℃下干燥10小时。磨碎经干燥的材料,并通过usp40-nf35第4552页在“hypermellose”下阐述的方法,以2wt%的水溶液测量“最终溶液粘度”。表1表1中的结果揭露将催化剂、增强剂和氧化剂中的全部三种组合在实现纤维素醚团的粘度降低方面的协同作用。compexi-l&exs2-15:催化剂+增强剂+氧化剂+纤维素醚的协同作用以timothythomson等人在us4845206中描述的方式使用适当的纤维素醚(参见表2)制备纤维素醚原料湿饼。·“a4m”对应于具有2663-4970mpa*s的粘度、27.5-31.5的甲氧基wt%的甲基纤维素(例如,methocela4m品牌的甲基纤维素醚)。·“e4m”对应于具有2663-4970mpa*s的粘度、28-30的甲氧基wt%和7-12的羟丙基wt%的羟丙基甲基纤维素(例如,methocele4m级羟丙基甲基纤维素)·“k4m”对应于具有2663-4970mpa*s的粘度、19-24的甲氧基wt%和4-12的羟丙基wt%的羟丙基甲基纤维素(例如,methocelk4m级羟丙基甲基纤维素)。将纤维素醚湿饼原料(以干基计为30千克;48wt%水分)装载到一立方米粒化机中,并在混合的同时添加硫酸铁和抗坏血酸的水溶液(每种物质的量参见表2)。混合30分钟。将所得混合物以恒定的速率与用水稀释的30%过氧化氢(如表2所示)并流进料到双螺杆配混机中。混合物在配混机中具有约三分钟的估计停留时间。离开配混机的纤维素醚具有较低的粘度,并且进入进料罐持续约5分钟,其从所述进料罐行进到冲击研磨机中以研磨干燥至低于5wt%的水分水平。表2中报告了干纤维素醚的所得粘度。compexsi和j证明了在不存在金属催化剂的情况下过氧化物解聚对最终产物粘度的影响。compexsk和l证明了在不存在抗坏血酸增强剂的情况下过氧化物解聚对最终产物粘度的影响。exs2-9证明了过氧化物解聚在一系列催化剂、增强剂和过氧化氢水平上对最终产物粘度的影响。数据还揭露了冲击研磨在从最终纤维素醚中去除氧化剂和水方面的有效性。exs10-15示出了在两种不同的过氧化物水平上增加抗坏血酸水平对最终纤维素醚粘度的影响。exs16-20:水增强的氧化剂去除实例16-20揭露了在冲击研磨之前添加水以便在干燥过程期间去除更多氧化剂的益处。将来自罗地格公司(德国帕德伯恩)的8升reaktordvt5rmk加热至60℃。45分钟后,在7分钟内在以75转/分钟用犁铧混合叶片混合的同时添加412.8克k4m纤维素醚(以上所述)和400克水(以四次100克剂量)。以恒定速度将所得的团混合60分钟。将0.55克硫酸铁和0.25克抗坏血酸溶解在150克水中,并在5分钟内在以恒定速度混合的同时将所得溶液喷到团上。混合额外25分钟。将团直接转移到来自werner&pfleiderer公司(德国丁克尔斯比尔(dinkelsbühl))的3.7升kneaderluk4-iii-1中,在添加前一小时将其加热至55℃。将团在55℃的捏合机中揉捏5分钟。制备25克的30%过氧化氢和水(4gex16、30gex17、60gex18、90gex19和120gex20)的溶液,并在揉捏的同时喷在团上。揉捏另外5分钟并且然后不揉捏静置10分钟。将团转移到冲击研磨装置(来自公司(德国哈姆(hamm))的ultra-rotor15(id2132))进行干燥和研磨。使用14,000转/分钟的研磨速度并设定温度,使得研磨机出口处的温度是110℃-120℃,其中氮气流量为40立方米/小时并且螺杆速度为10转/分钟。干燥400克团,并丢弃以清洁装置。干燥剩余的团样品并且然后通过以下溶液过氧化物评估来表征其残留的过氧化氢:制备2wt%的纤维素醚水溶液并且然后使用商业淀粉-碘测试条(mqant0.5-25ppm)通过将测试条浸入水溶液中一秒钟并且移出来确定过氧化氢浓度。90秒后,将测试条的颜色与来自制造的颜色标准进行比较。表3含有在处理团之前添加到30%过氧化氢中的水量以及所得的过氧化氢浓度。将过氧化氢浓度确定为纤维素醚团的2wt%水溶液中的浓度。表3样品ex16ex17ex18ex19ex20添加的水(g)4306090120过氧化氢(ppm)2510555表3中的数据揭露了在冲击研磨之前添加水有助于在冲击研磨期间消除氧化剂。溶液相筛选工作在纤维素醚溶液上进行工作以筛选合适的催化剂、增强剂和氧化剂。在溶液相筛选工作中,降解反应是在小规模的纤维素醚溶液中进行的,而不是在纤维素醚团上进行的。预期溶液中的性能反映纤维素醚团(如在本发明的配混步骤期间形成的)中的性能,因为化学过程是相同的。对于溶液相筛选工作,制备纤维素醚溶液,并将添加剂添加到溶液中。然后将溶液密封到具有延伸到溶液中的搅拌桨的容器中,并通过电动机以恒定速率搅拌所述桨。监测施加到电动机上以搅拌溶液中的桨的电流。电流与搅拌溶液所需的力成比例,所述力与溶液的粘度成比例。因此,施加到桨的电动机的电流与溶液的粘度成比例。将装置校准为各种粘度标准并且确定将施加的电流转换为溶液粘度的转换系数。因此,通过监测施加的电流,监测溶液的粘度。实验确定了4000mpa*s的原始粘度到2000mpa*s所需的时间量(此处指定为“降解半衰期”)。与这些类似的结果还可以通过以下方式获得:在haakeviscotesteriq中使用0.2nmm-100nmn的扭矩范围、0.1rpm至1500rpm的转速、珀耳帖(peltier)温度模块tm-pe-c,haake转子fl26用“connectassist”微芯片、以及用于tm-pe-c的haakecupcb25din运行筛选反应的同时监测溶液粘度。在确定反应组合物在反应期间是否倾向于将颜色引入纤维素醚的努力中还监测了溶液的变色。变色通过紫外/可见(uv/vis)光谱、l*ab颜色和δeab变色监测。在shimadzuuv-3600uv/vis/nir光谱仪上使用1厘米x1厘米丙烯酸一次性比色皿测量uv/vis光谱。以5纳米间隔记录380到780纳米的吸收。使用astmee308标准使用cie19312°标准观察者三色刺激值和模拟的d65标准照明源从卷积积分计算颜色坐标,并且如同一标准中所述的将所得xyz坐标转换为l*ab颜色坐标。通过公式δeab=((100-l*)2+a2+b2)1/2从l*ab色值确定δeab变色值。催化剂储备溶液根据表4制备基于金属阳离子或催化物种的50毫摩尔(mm)浓度的催化剂候选物的储备溶液。表4候选物mw(g/mol)量(mg)μ摩尔盐/μ摩尔活性添加的水(ml)feso4*7h2o278.01139.0500/50010fe(so4)3399.88100.0250/50010cuso4*5h2o249.69125.9500/50010kmno4158.0379.0500/50010ki166.0083.0500/50010tio2(21nm部分)79.8739.9500/50010zno(<50nm部分)81.4140.7500/50010氧化剂储备溶液表5列出了氧化剂候选物。表5芬顿增强剂储备溶液制备了以下试剂的浓度为1m的20ml储备溶液。(1)抗坏血酸-用naoh缓冲至ph2(校准的ph计),缩写为“asc-ph2”,(2)抗坏血酸-用naoh缓冲至ph5,缩写为“asc-ph5”,(3)柠檬酸-用naoh缓冲至ph2,缩写为“cit-ph2”,(4)柠檬酸-用naoh缓冲至ph5,缩写为“cit-ph5”,(5)过硫酸钠,缩写为“过硫酸盐”,(6)葡萄糖,(7)偏亚硫酸氢钾,缩写为“亚硫酸氢盐”或“metabisulf.”,(8)异抗坏血酸,缓冲至ph5,缩写为“ery-ph5”,(9)硫代硫酸钠,缩写为“硫代硫酸盐”。在实验之间,将储备溶液冷藏。螯合剂储备溶液通过在20ml水中搅拌适量的edta-na2制备20ml的edta-na2在水(50mm)中的储备溶液。酸/碱按购买的原样使用0.1m氢氧化钠和0.05m硫酸溶液。纤维素醚储备溶液如下制备methoceltme4m品牌纤维素醚的储备溶液:将735ml的18.2mω·cm-1的水加热至即将沸腾的状态并且添加15g的methoceltme4m品牌纤维素醚。将悬浮液通过顶置式搅拌器剧烈搅拌直至纤维素醚完全悬浮并且没有结块留下。继续缓慢地(约20rpm)搅拌溶液,并使其冷却至室温。将纤维素醚溶液等分试样(20g)分配到30mlvicar玻璃小瓶中。淬灭剂储备溶液对于潜在的化学计量猝灭剂,制备了20ml的浓度为1n的储备水溶液。(1)1m抗坏血酸-用naoh缓冲至ph=2,(2)1m抗坏血酸-用naoh缓冲至ph=5,(3)1m异抗坏血酸-用naoh缓冲至ph=5,(4)1m次磷酸钠,(5)1m脲,(6)0.2m单宁酸,(7)1m半胱氨酸,(8)0.5m偏亚硫酸氢钾,(9)1m硫代硫酸钠,(10)1m蔗糖,(11)1mdmso,(12)柠檬酸,用naoh缓冲至ph=5。(13)按购买的原样使用次氯酸钠溶液(“4%-5%活性cl”≈0.634mnaocl)。如下制备潜在的催化或芬顿催化剂降解淬灭剂:(1)在水中的10mm碘化钠,(2)在水中的50mmedta-na2。通过将1.25mg至20mg的冻干物(奥德里奇公司(aldrich))溶解在10ml的微滤磷酸盐缓冲液(10mm,ph=7.0)中,以250u/ml至10000u/ml的浓度新鲜制备牛过氧化氢酶储备溶液。按从mp生物医疗公司(mpbiomedicals)接收的原样使用黑曲霉过氧化氢酶(≥1000u/ml的溶液)。贝克(baker)过氧化氢测试条贝克过氧化氢测试条可从jt贝克公司(jtbaker)获得并且可以与具有1至100mg/l的过氧化氢检测范围的其他可商购过氧化氢测试互换使用。阴性过氧化物浸入测试结果意指测试溶液含有低于1mg/l的过氧化氢(如通过使用测试条进行浸入测试确定的)。催化剂筛选进行如表6中列出的48个实验,对于每个实验,使用以下程序。向玻璃小瓶中添加2wt%的methoceltme4m溶液的20克(g)等分试样,并且然后在适用的情况下添加催化剂储备溶液(100微升,对应于5微摩尔活性催化剂),然后添加氢氧化钠(0.1n)或硫酸(0.1n)溶液作为“ph调节剂”(50微升,对应5微摩尔质子或氢氧根阴离子)(在指示时)。以300rpm搅拌反应5分钟,其后通过注射器添加30%过氧化氢(400微升,用蒸馏水稀释至1毫升,约3.92mmol)。在以300rpm混合的同时并且在25℃下运行反应持续3小时。结果记录在表6中并绘制在图1和2中。导致较短的降解半衰期(4000mpa*s降解至2000mpa*s的较少时间)的催化剂候选物,反应更快并且催化剂更令人希望。如图1中明显的,硫酸铁(ii)、硫酸铁(iii)、硫酸铜(ii)和氧化锌(ii)具有催化作用,因为它们导致比没有催化剂候选物的空白参照物短的半衰期。图2还揭露了相对于没有催化剂的空白参照物,硫酸铁(ii)、硫酸铁(iii)、硫酸铜(ii)和氧化锌(ii)都没有导致进一步的变色。表6增强剂筛选进行如表7中列出的56个实验,对于每个实验,使用以下程序。向玻璃小瓶中添加2wt%的methoceltme4m溶液的20克(g)等分试样,并且然后在适用的情况下添加催化剂储备溶液(100微升,对应于5微摩尔活性催化剂),然后添加氢氧化钠(0.1n)或硫酸(0.1n)溶液作为“ph调节剂”(50微升,对应5微摩尔质子或氢氧根阴离子)(在指示时)。在指示时,添加edta(50毫摩尔储备溶液;100微升,对应于5微摩尔edta-na2),然后添加芬顿增强剂(一摩尔储备溶液;200微升(用蒸馏水稀释至一毫升),约3.92毫摩尔)。以300rpm搅拌5分钟并且然后通过注射器添加h2o230%(400微升,用蒸馏水稀释至1毫升,约3.92毫摩尔)。在没有催化剂的情况下运行空白反应,并且添加去离子水而不是过氧化氢溶液。反应在300rpm和25℃下运行3小时。结果在表7和图3-图6中。图3和图4示出使用硫酸铁(iii)催化剂的结果。图3示出抗坏血酸和偏亚硫酸氢钠提高了反应速率。其也揭露了包含edta减慢反应速度。图4还示出对于具有和不具有edta的样品,抗坏血酸、过硫酸钠和偏亚硫酸氢钠都改善了颜色。图5和图6示出使用硫酸酮(iii)催化剂的结果。图5示出抗坏血酸将反应速率提高了大于两个数量级。图6还示出抗坏血酸、过硫酸钠和偏亚硫酸氢钠改善颜色,特别是对于不具有edta的样品。表7氧化剂筛选进行如表8中列出的36个实验,对于每个实验,使用以下程序。向玻璃小瓶中添加2wt%的methoceltme4m溶液的20克(g)等分试样,并且然后在适用的情况下添加催化剂储备溶液(100微升,对应于5微摩尔活性催化剂),然后添加氢氧化钠(0.1n)或硫酸(0.1n)溶液作为“ph调节剂”(50微升,对应5微摩尔质子或氢氧根阴离子)(在指示时)。在指示时,添加芬顿增强剂(一摩尔储备溶液;100微升)。以300rpm搅拌5分钟,并且然后通过注射器添加如表8中所示的储备氧化溶液(h2o2150微升;过乙酸310微升;过硫酸钠735微升;全部用蒸馏水稀释至1毫升,约1.47毫摩尔氧化剂)。在没有催化剂的情况下运行空白反应,并且添加去离子水而不是过氧化氢溶液。反应在300rpm和25℃下运行3小时。结果在表8和图7-图10中。图7和图8示出使用硫酸铁(iii)催化剂的结果。图7示出了抗坏血酸和异抗坏血酸普遍减少了反应的降解半衰期,而偏亚硫酸氢钠和硫代硫酸钠对于所选氧化剂减少了降解半衰期。图8揭露了抗坏血酸和异抗坏血酸仅对于过氧化氢改善颜色,而偏亚硫酸氢钠和硫代硫酸钠对于所有氧化剂改善颜色。图9和图10示出使用硫酸酮(ii)催化剂的结果。图9揭露了抗坏血酸和异抗坏血酸对于所有氧化剂都减少了降解半衰期,而偏亚硫酸氢钠和硫代硫酸钠对于一些氧化剂改善降解半衰期。表8淬灭剂筛选(a)淬灭剂筛选i.将2wt%methocele4m纤维素醚溶液的20g等分试样添加到七个单独的玻璃小瓶中。在第八个玻璃小瓶中添加水作为空白样品。添加硫酸铁(iii)储备溶液(100微升,对应于5微摩尔活性催化剂),然后添加硫酸溶液(0.1n)(50微升,对应于5微摩尔质子)。在阴性对照小瓶和空白样品中没有添加催化剂。以300rpm搅拌反应5分钟并且然后通过注射器添加30%h2o2(400微升,用蒸馏水稀释至1毫升,约3.92毫摩尔),除了阴性对照小瓶和空白样品外。20分钟后,将淬灭测试溶液(1.5毫升以下项之一:1m脲水溶液;0.2m单宁酸,1m半胱氨酸,0.5m偏亚硫酸氢钾,0.01m碘化钠,1m硫代硫酸钠)添加到单独的小瓶中,除了阴性和阳性对照小瓶以及空白样品外。记录粘度降解持续三个小时。与空白、阴性和阳性对照样品相比的碘化钠样品的降解曲线示于图13中。通过使用贝克过氧化氢测试条进行浸入测试来测量h2o2含量。偏亚硫酸氢盐、硫代硫酸盐、半胱氨酸和阴性对照小瓶的过氧化物测试条为阴性(指示成功淬灭),而其他样品的过氧化物测试条为阳性。(b)淬灭剂筛选ii.将2wt%methocele4m纤维素醚溶液的20g等分试样添加到七个单独的玻璃小瓶中。将水添加到第八个小瓶中作为空白样品。然后添加硫酸铁(iii)储备溶液(10微升,对应于5微摩尔活性催化剂),然后添加硫酸溶液(0.1n)(50微升,对应于5微摩尔质子)。在阴性对照小瓶和空白样品中没有添加催化剂。以300rpm搅拌反应5分钟并且然后通过注射器添加30%h2o2(400微升,用蒸馏水稀释至1毫升,约3.92毫摩尔),除了阴性对照小瓶和空白样品外。20分钟后,向每个小瓶中添加淬灭测试溶液(1.5ml的:(a)1m蔗糖;(b)1m次磷酸钠;(c)1m在水中的二甲基亚砜;(d)50mmedta-na2;或(e)2.0ml次氯酸钠溶液(约0.63mnaocl))到单独的小瓶中,除了阴性和阳性对照以及空白样品外。记录粘度降解持续三个小时。与空白样品、阴性和阳性对照样品相比的蔗糖、二甲基亚砜、edta-na2和dmso的降解曲线示于图13中。用贝克过氧化氢测试条测量h2o2含量。过氧化物测试条对于次氯酸盐为阴性,对于次磷酸盐减弱并且对于dmso显著减弱,并且对于蔗糖和edta-na>100mg/l。(c)淬灭剂筛选iii.使用作为淬灭测试溶液的(a)1.5ml抗坏血酸(1m,用naoh缓冲至ph为2),(b)1.5ml抗坏血酸(1m,用naoh缓冲至ph为5),(c)1.5ml异抗坏血酸(1m,用naoh缓冲至ph为5),(d)次磷酸钠(1m)和(e)柠檬酸(1m,用naoh缓冲至ph为5)运行与先前的筛选类似的筛选。过氧测试条对于抗坏血酸(在ph2和5下)和异抗坏血酸均为阴性。测试条对于次磷酸盐和柠檬酸为>100mg/l。(d)过氧化氢酶淬灭剂:系列i.通过将10毫克牛过氧化氢酶(奥德里奇公司)溶解在5毫升冷的微过滤的磷酸钠缓冲液(50mm,ph7)中来制备新鲜的过氧化氢酶储备溶液,产生具有4,000-10,000u/ml活性的溶液(按照制造商提供的规格)。用额外的磷酸盐缓冲液将此溶液的1.5ml等分试样稀释至6ml的总体积,产生1,000-2,500u/ml过氧化氢酶储备溶液。从第二种储备溶液中,进一步用磷酸盐缓冲液稀释1.5ml等分试样至6ml的总体积,产生250-625u/ml溶液。将溶液储存在冰箱中直至使用。向具有2wt%methocele4m溶液的20g等分试样的玻璃小瓶中添加硫酸铁(iii)储备溶液(100微升,对应于5微摩尔活性催化剂),然后添加硫酸溶液(0.1n)(50微升,对应于5微摩尔质子)。阴性对照小瓶中没有添加催化剂。以300rpm搅拌反应5分钟并且然后通过注射器添加30%h2o2(400微升,用蒸馏水稀释至1ml,约3.92mmol),除了阴性对照小瓶外。20分钟后,将碳酸钠缓冲液(2ml,500mm)添加到三个小瓶中。添加这三种牛过氧化氢酶溶液的1ml等分试样,一种添加到无磷酸盐缓冲液的三个小瓶中的每一个中,并且每种之一添加到具有磷酸盐缓冲液的三个反应小瓶中。阴性和阳性对照反应中没有添加过氧化氢酶。监测所有反应的粘度降解持续3小时,之后通过用贝克过氧化氢测试条进行浸入测试来测量h2o2含量。含过氧化氢酶的反应不含任何剩余的过氧化氢。(e)过氧化氢酶淬灭剂:系列ii.通过将5mg牛过氧化氢酶溶解在10ml冷的微过滤的磷酸钠缓冲液(50mm,ph7)中来制备新鲜的过氧化氢酶储备溶液,产生1,000-2,500u活性(按照制造商规格)。将储备溶液保持在5℃直至使用。在六个玻璃小瓶中制备测试溶液。向小瓶1和2中添加2wt%methocele4m溶液的20g等分试样和硫酸铁(iii)储备溶液(100微升,对应于5微摩尔活性催化剂)和硫酸溶液(0.1n)(50微升,对应于5微摩尔质子)和1摩尔异抗坏血酸水溶液(用氢氧化钠缓冲至ph5)(100微升)。向小瓶3和4中添加2wt%methocele4m的20g等分试样和硫酸铜(ii)储备溶液(100微升,对应于5微摩尔活性催化剂)、异抗坏血酸水溶液(用氢氧化钠缓冲至ph5)(100微升)。向小瓶5和6中,添加2wt%methocele4m的20g等分试样和硫酸铜(ii)储备溶液(100微升,对应于5微摩尔活性催化剂),然后添加氢氧化钠溶液(0.1n)(50微升,对应于5微摩尔氢氧根阴离子)。监测所有小瓶的粘度降解反应持续3小时,之后通过用贝克过氧化氢测试条进行浸入测试来测量h2o2含量。所有含过氧化氢酶的反应均不含任何剩余的过氧化氢。纤维素醚降解的并排比较为了示出所要求保护的本发明的反应动力学的改善,使用溶液相筛选技术进行了若干氧化方法。对于每个反应,将20g的2wt%methocele4m溶液放入小瓶中并搅拌5分钟。然后,添加下述添加剂,并记录小瓶中溶液的随时间推移的溶液粘度变化。空白.对于空白样品,将溶液搅拌5分钟并且然后将200微升水添加到溶液中,并将溶液搅拌额外3小时。仅过氧化氢.对于仅过氧化氢运行,将溶液搅拌5分钟并且然后将4mmolh2o2添加到纤维素醚溶液中,并将溶液搅拌3小时,监测粘度变化。过氧化氢/硫酸铁.对于过氧化氢/硫酸铁运行,将溶液搅拌5分钟并且然后添加2.5微摩尔硫酸铁(iii),并将所得溶液搅拌5分钟并且然后添加4毫摩尔h2o2,并将溶液搅拌3小时,监测粘度变化。过氧化氢/硫酸铁/抗坏血酸运行.对于过氧化氢/硫酸铁/抗坏血酸运行,将溶液搅拌5分钟并且然后将2.5微摩尔的硫酸铁(iii)和200微摩尔的抗坏血酸添加到溶液中,并将所得溶液搅拌5分钟并且然后添加4毫摩尔的h2o2,并将溶液搅拌3小时,监测粘度变化。减少载荷-过氧化氢/硫酸铁/抗坏血酸运行.对于此运行,除使用100微摩尔抗坏血酸外,与上一运行相同地进行。来自这些运行的结果绘制在图11中,所述图示出溶液粘度随时间的变化。结果表明具有所要求保护的本发明的组分的溶液的粘度急剧下降。硫酸铜(ii)运行.使用硫酸铜(ii)代替硫酸铁(iii)重复以上最后三种运行。结果绘制在图12中并且示出在本发明的上下文中硫酸铜(ii)配制品也产生粘度的急剧下降。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1