一种以哒嗪衍生物为配体的双环金属铂(II)配合物及其制备方法和应用与流程
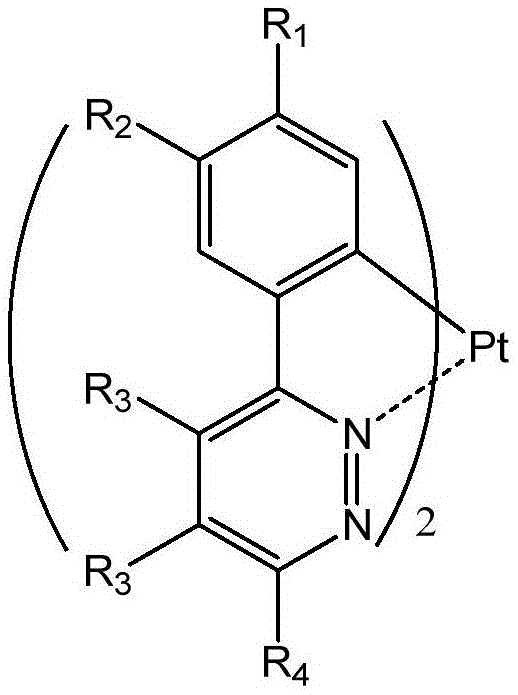
本发明属于有机光电材料技术领域,具体地说,涉及一种以哒嗪衍生物为配体的双环金属铂(ii)配合物及其制备方法和应用。
背景技术:
有机电致发光二极管(oled)由于同时具备自发光、不需背光源、对比度高、厚度薄、视角广、反应速度快、可用于挠曲性面板、使用温度范围广、构造及制程较简单等优异的特性,被认为是下一代的平面显示器新兴技术。从oled使用的有机发光材料来看,磷光金属配合物由于过重原子效应,其电致发光器件的内量子效率理论上可以达到100%,而在这些磷光配合物中,环金属铂(ii)配合物是广泛研究的配合物之一,拥有效率高、激发态寿命短、颜色可调性强等优点。
环金属铂(ii)配合物有单环金属铂(ii)配合物和双环金属铂(ii)配合物两种,其中双环金属铂(ii)配合物有更多的c-金属键,稳定性更强,且发光效率也更高,由于较高的稳定性,基于此类材料的oled器件的使用寿命也更长。但双环金属铂(ii)配合物的合成难度非常大,文献中报道的单环金属铂(ii)配合物较多,但双环金属铂(ii)配合物则非常少,甚至几乎查不到双环金属铂发光性能的报道。目前,双环金属铂(ii)配合物普遍采用的制备方法有如下四种:
(1)将配体生成锂盐提高反应活性后与铂盐配位(inorganicchemistry,1984,23,4249-4253;inorganicchemistry,1987,26,2814-2818;helveticachimicaacta,1989,72,377-382;inorganicchemistry,1996,35,4883-4888,inorganicchemistry,1996,35,4889-4895),由于配体生成锂盐需要无水、无氧及低温条件,条件非常苛刻,且脱氢的位置也很难控制,很多官能团(如酯基,硝基以及含活波氢的官能团)都不能耐受这样的条件,因此该方法的应用很受限制;
(2)采用活性非常高的二价铂化合物为原料来制备,例如,[pt2(ch3)4(μ-s(ch3)2)2]这种二价铂配合物中由于有反应活性强的甲基,可以夺取配体上的氢,使配体易于跟铂配位,但是这种铂原料的制备条件同样苛刻,产率也较低,因此非常昂贵(journaloforganometallicchemistry,2013,723,188-197);在用[pt(c6h4ch2nme2)cl]2(angewandtechemieinternational.edition,1994,33,783-784)做活性高的铂原料时,甚至还需要用到银催化剂agbf4来提高反应产率,而银催化剂同样也比较贵;
(3)用环金属铂配合物原料与配体在催化剂存在下反应生成目标双环金属铂配合物(chemistryaeuropeanjournal,2013,19,16722-16728),催化剂一般为高活性的银盐(如agbf4)或碱,该方法要通过两步反应才能得到最终的双环金属铂配合物,反应步骤较长,产率波动也同样较大,同时环金属铂配合物原料同样难以获得;
(4)配体与价格较高的油溶性二价铂盐[pt(dmso)2cl2]在混合二甲苯中,碳酸钠存在下回流来合成(chemistryaeuropeanjournal,2013,19,16722-16728),该方法温度较高,且回流时间为两到四天,报道的两种配合物的产率分别为5%和63%,因此不同配体用该方法反应时产率波动较大,且未见该方法用于其它配体来制备双环金属铂配合物的报道。同时由于配体都是平面刚性的结构,导致这些配合物的堆积严重,不适合作为发光材料使用。实际上文献也未报道其发光性能。
我们知道,在一般的环金属配体结构中,与金属配位的n原子的邻位为c原子,c原子上有个h原子,而在哒嗪类环金属配体中,与金属配位的n原子的邻位为n原子,而非h原子,免除了h原子干扰,有利于配体与金属配位,也就是说哒嗪类配体的配位能力比其它配体要强,其结果是使其易于生成双环金属铂配合物,且生成产物的配位键更短,从而增加了配合物的稳定性。而且哒嗪类配体的反应活性可以通过引入取代基进一步提升,例如我们将含氯的苯基酞嗪衍生物为环金属化配体合成了三环铱配合物ir(mpcppz)3,发现了该配体与三氯化铱通过一步反应直接高产率生成三环铱配合物的独特现象(journalofmaterialschemistry,2008,18,1636-1639)。但是在铂配合物中是否也能利用哒嗪类配体反应活性高的性质,在文献中并没有报道。而且与铱配合物的球性构型不同,铂配合物为平面构型,单环金属化配位的中间产物容易在反应过程中聚集沉淀,难以继续与配体反应得到双环金属化的产物,同时二价铂离子为不稳定的中间价态,容易被配体氧化或还原,不易得到双金属化配合物。因此在配体反应活性高的条件下,还需要降低配合物中间产物的聚集沉淀,降低配体的氧化还原活性。
技术实现要素:
1.要解决的问题
针对现有制备双环金属铂(ii)配合物需昂贵的原料、合成路线繁琐、重复性差以及产物发光性能不理想的问题,本发明提供一种以哒嗪衍生物为配体的双环金属铂(ii)配合物及其制备方法和应用,本发明制备方法用哒嗪衍生物作为配体,提高配体与铂的反应活性,使其能与二价铂盐通过一步反应高产率生成哒嗪类双环金属铂(ii)配合物。
2.技术方案
为了解决上述问题,本发明所采用的技术方案如下:
一种以哒嗪衍生物为配体的双环金属铂(ii)配合物,该配合物的结构通式如下所示:
其中,r1、r2为卤素原子、卤代烷基或h原子;r3为烷基、取代芳烷基或取代芳基;r4为烷氧基、烷硫基、取代芳氧基、取代芳硫基、取代胺基、取代芳胺基或取代芳基;并且上述烷基和取代基为1~10个碳原子的烷基。
更进一步地,所述的r1、r2为卤素原子、卤代烷基时,卤素原子为氟、氯或溴原子中的一种。
更进一步地,所述的r3为烷基,取代芳烷基或取代芳基时,芳烃为苯、萘、联苯中的一种。
更进一步地,所述的r4为取代芳氧基、取代芳硫基、取代胺基、取代芳胺基或取代芳基时,芳烃为苯、萘、联苯中的一种。
上述的一种以哒嗪衍生物为配体的双环金属铂(ii)配合物的制备方法,包括以下步骤:
(1)以3,6-二氯哒嗪衍生物、芳基硼酸或硼酸酯、碱为原料,在催化剂作用下,合成哒嗪类配体;
(2)将哒嗪类配体与二价铂盐、溶剂及碱混合,得到所述的以哒嗪衍生物为配体的双环金属铂(ii)配合物。
更进一步地,所述的步骤(1)中将3,6-二氯哒嗪衍生物、芳基硼酸或硼酸酯、碱溶于溶剂中,在催化剂作用下,60~150℃进行偶合反应,反应2~24h,得到a类哒嗪类配体;其中,反应物用量按摩尔分数计:3,6-二氯哒嗪衍生物1份,芳基硼酸或硼酸酯2~4份,碱1~6份,溶剂50~500份,催化剂0.01~1份;所述的碱为碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、氢氧化钡、醋酸钠、碳酸铯中的一种;所述的溶剂为四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、水、甲苯或者其混合物中的一种;所述的催化剂为钯催化剂,包括pd(pph3)4、pd(pph3)2cl2、pd(dppf)cl2。反应过程如下所示:
更进一步地,所述的步骤(1)包括步骤(1-1)和步骤(1-2),具体步骤如下:
(1-1)将3,6-二氯哒嗪衍生物、芳基硼酸或硼酸酯、碱溶于溶剂中,在催化剂作用下,60~150℃进行偶合反应,反应2~24h,得到单氯取代哒嗪类中间体;反应过程如下所示:
(1-2)将单氯取代哒嗪类中间体、芳基硼酸或硼酸酯、碱溶于溶剂中,在催化剂作用下,60~150℃进行偶合反应,反应2~24h,得到b类哒嗪类配体;或者将单氯取代哒嗪类中间体与含活性氢的化合物、碱混合,在有机溶剂中,0~150℃反应2~24h,得到c类哒嗪类配体;反应过程分别如下所示(其中,rb为氢原子或烷基中的一种,rb的个数可以为1~5个中的任意一种。rc为烷基、取代芳基中的一种,芳基为苯、萘、联苯中的一种,取代基的个数为1~5个中的任意一种。x为氧原子、硫原子或氮原子。):
其中,所述步骤(1-1)中反应物用量按摩尔分数计:3,6-二氯哒嗪衍生物1份,芳基硼酸或硼酸酯0.5~1.5份,碱1~6份,溶剂50~500份,催化剂0.01~1份;所述的步骤(1-2)中合成b类哒嗪类配体时,反应物用量按摩尔分数计:将单氯取代哒嗪类中间体1份,芳基硼酸或硼酸酯1~2份,碱1~6份,溶剂50~500份,催化剂0.01~1份;另外,所述步骤(1-1)中和所述的步骤(1-2)中合成b类哒嗪类配体时,所述的碱为碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、氢氧化钡、醋酸钠、碳酸铯中的一种;所述的溶剂为四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、水、甲苯或者其混合物中的一种;所述的催化剂为钯催化剂,包括pd(pph3)4、pd(pph3)2cl2、pd(dppf)cl2。
更进一步地,所述步骤(1-2)中合成c类哒嗪类配体时,反应物用量按摩尔分数计:单氯取代哒嗪类中间体1份,含活性氢的化合物1~3份,碱1~6份,有机溶剂50~500份;所述的含活性氢的化合物为醇、酚、硫酚或芳胺;所述的碱为正丁基锂、二异丙基胺锂、氢化钠、碳酸钠、碳酸钾、氢氧化钠、氢氧化钠、碳酸铯中的一种;所述的有机溶剂为四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、乙腈、甲苯或者其混合物中的一种。
更进一步地,所述步骤(2)中将哒嗪类配体和二价铂盐,在溶剂中,50~150℃反应5~24h,得到以哒嗪衍生物为配体的双环金属铂(ii)配合物;其中,反应物用量按摩尔分数计:哒嗪类配体2~4份,二价铂盐1份,溶剂50~500份;所述的二价铂盐为二氯化铂、四氯铂酸钾、四氯铂酸钠、四氯铂酸锂、四氯铂酸铵、二氯二(二乙基硫醚)铂,二氯二(二甲亚砜)铂;所述的溶剂为水、甲氧基乙醇、乙氧基乙醇、乙二醇、丙二醇、甘油、缩水甘油醚或者其混合物中的一种。
上述的一种以哒嗪衍生物为配体的双环金属铂(ii)配合物在有机电致发光二极管领域中的应用。
本发明的哒嗪衍生物配体含有氟烷基、环烷基、多烷基苯等有位阻的取代基,能够增加单环金属化配位的反应中间体的溶解性,促进双环金属铂配合物的生成,同时由于配体的反应活性高可以在较低温度下进行配位反应,降低了反应温度,能有效减少铂的氧化还原几率,能够有效地降低材料的制备成本;而且位阻基团的存在能有效降低铂配合物由于分子间堆积产生的发光淬灭,提高材料的稳定性和发光性能,增加其实际应用的价值。
3.有益效果
相比于现有技术,本发明的有益效果为:
(1)本发明提供的双环金属铂(ii)配合物的制备方法采用哒嗪衍生物作为配体,且通过卤素原子修饰,使配体具有更大的反应活性,在与二价铂盐反应时,能在温和条件下,直接一步反应生产具有发光性能的双环金属铂(ii)配合物,且产率在50%以上;
(2)本发明提供的双环金属铂(ii)配合物的制备方法采用的铂原料的价格便宜,是最常见的二价铂盐,能有效降低制备成本;
(3)本发明提供的双环金属铂(ii)配合物稳定性高,质量减少5%的热失重温度在350℃以上,且不易发生分子聚集,光致发光量子效率在60%以上。
附图说明
图1为本发明以哒嗪衍生物为配体的双环金属铂(ii)配合物的结构通式;
图2为本发明实施例1中双环金属铂(ii)配合物a在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
图3为本发明实施例2中双环金属铂(ii)配合物b在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
图4为本发明实施例3中双环金属铂(ii)配合物c在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
图5为本发明实施例4中双环金属铂(ii)配合物d在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱。
图6为本发明实施例5中双环金属铂(ii)配合物e在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱。
具体实施方式
下面结合具体实施例对本发明进一步进行描述。
实施例1
将2.3g(10mmol)3,6-二氯-4,5-环己(1’,4’)基哒嗪、3.8g(20mmol)间三氟甲基苯硼酸和1.4g(10mmol)碳酸钾加入250ml圆底烧瓶中,加入0.2g(0.3mmol)pdcl2(dppf),再倒入30ml四氢呋喃和15ml水,n2保护,100℃回流12h。冷却,旋干四氢呋喃,用二氯甲烷萃取,得下层有机相,旋干,用石油醚:乙酸乙酯=1:1洗脱剂硅胶柱层析分离,得4.1g3,6-二(间三氟甲基苯基)-4,5-环己(1’,4’)基哒嗪的白色固体,产率为91%。产物的核磁图谱为:1hnmr(400mhz,cdcl3)δ8.00(s,2h),7.92(d,j=7.6hz,2h),7.82(d,j=7.8hz,2h),7.72(t,j=7.7hz,2h),3.43(s,2h),1.98(d,j=7.9hz,4h),1.52(d,j=7.5hz,4h)。19fnmr(cdcl3,367mhz)δ-62.86(s,6f)。产物的质谱为:ms((+)-esi):m/z=449.67(calcd.449.14forc24h19f6n2,[m+h+])。
将1.8g(4mmol)3,6-二(间三氟甲基苯基)-4,5-环己(1’,4’)基哒嗪、四氯铂酸钾0.8g(2mmol),乙氧基乙醇45ml和水15ml置于120ml的封管中,在n2保护下,120℃下反应12h。反应结束后冷却至室温,用布氏漏斗进行抽虑得到黄色固体,再用二氯甲烷进行硅胶柱层析,得到配合物a的黄色固体1.3g,产率为60%。产物的核磁图谱为:1hnmr(400mhz,cdcl3)δ7.93(s,2h),7.56(d,j=6.3hz,2h),7.42(s,2h),7.34(t,j=8.1hz,2h),7.24(d,j=9.6hz,6h),4.04(s,2h),3.02(s,2h),1.58(d,j=19.8hz,8h),1.24(s,8h)。19fnmr(cdcl3,367mhz)δ-61.59(s,6f),-62.67(s,6f)。产物的质谱为:ms((+)-esi):m/z=1090.2319(calcd.1090.2318forc48h35f12n4pt,[m+h+])。产物在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱图如图2所示,该配合物的吸收峰位λmax=291nm,发射峰位λmax=510nm。发光量子效率为89%,5%的热失重温度为396℃。产物的结构式如下所示:
实施例2
将10g(68mmol)3,6-二氯哒嗪,12.8g(68mmol)间三氟甲基苯硼酸和9.38g(68mmol)碳酸钾加入500ml圆底烧瓶中,加入[1,1,-双(二苯基膦基)二茂铁]二氯化钯(pdcl2(dppf))1.1g(1.6mmol),再倒入45ml四氢呋喃和30ml水,n2保护,回流12h。冷却,旋干四氢呋喃,用二氯甲烷溶液萃取,有机相旋干,用石油醚:乙酸乙酯=3:1洗脱剂硅胶柱层析分离,得10.2g3-(3-三氟甲基苯基)-6-氯哒嗪的白色固体,产率为58%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ8.31(s,1h),8.26(d,j=7.8hz,1h),7.88(d,j=9.0hz,1h),7.78(d,j=7.8hz,1h),7.67(t,j=7.8hz,1h),7.62(d,j=9.0hz,1h);19fnmr(376mhz,cdcl3)δ-62.66(s,3f).ms((+)-esi):m/z=259.02(calcd.259.03forc11h7clf3n2,[m+h+])。
将5g(19mmol)3-(3-三氟甲基苯基)-6-氯哒嗪、6g(38mmol)2,4,6-三甲基苯硼酸和5g(38mmol)碳酸钾加入500ml圆底烧瓶中,加入0.6g(5wt%)pdcl2(dppf),再倒入45ml四氢呋喃和30ml水,n2保护,回流12h。冷却,旋干thf,用二氯甲烷溶液萃取,得下层有机相,旋干,用石油醚:乙酸乙酯=6:1洗脱剂硅胶柱层析分离,得2g3-(3-三氟甲基苯基)-6-(2,4,6-三甲基苯基)哒嗪的白色固体,产率为30%。产物的核磁数据为:1hnmr(400mhz,cdcl3)δ8.45(s,1h),8.37(s,1h),7.97(d,j=13.5hz,1h),7.77(d,j=7.8hz,1h),7.68(t,j=7.8hz,1h),7.49(d,j=7.0hz,1h),7.00(s,2h),2.36(s,3h),2.08(s,6h);19fnmr(377mhz,cdcl3)δ-62.77(s,3f)。产物的质谱数据为:ms((+)-esi):m/z=343.33(calcd.343.13forc20h18f3n2,[m+h+])。
将1.4g(4mmol)3-(3-三氟甲基苯基)-6-(2,4,6-三甲基苯基)哒嗪、四氯铂酸钾0.8g(2mmol),乙氧基乙醇45ml和水15ml置于120ml的封管中,在n2保护下,120℃下反应12h。反应结束后冷却至室温,用布氏漏斗进行抽虑得到黄色固体,再用二氯甲烷进行硅胶柱层析,得到配合物b的黄色固体1.1g,产率为66%。配合物b的核磁数据为1hnmr(400mhz,cdcl3)δ7.99(d,j=9.0hz,2h),7.80(s,2h),7.26-7.17(m,6h),6.72(s,4h),2.24(s,6h),1.57(s,6h),1.24(s,6h);19fnmr(377mhz,cdcl3)δ-61.72(s,6f)。产物的质谱数据为:ms((+)-esi):m/z=878.2260(calcd.878.2257forc40h33f4n4pt,[m+h+])。配合物b在二氯甲烷中形成的稀溶液的紫外可见吸收光谱和光致发光光谱如图3所示,该配合物的吸收峰位λmax=286nm,发射峰位λmax=508nm。另外,发光量子效率为77%,5%的热失重温度为386℃。产物的结构式如下所示:
实施例3
将3-(3-三氟甲基苯基)-6-氯哒嗪1.3g(5mmol),2,4,6-三甲基苯酚0.8g(6mmol)置于120ml的反应瓶中,向反应瓶中加入碳酸钾1.4g(10mmol),将n,n-二甲基甲酰胺50ml作为溶剂,向反应瓶中通入n2密封保护,反应回流12h,反应结束冷却至室温,用乙酸乙酯进行萃取得有机相,并用水对有机相进行反萃取,有机相旋干后用石油醚和乙酸乙酯比为5:1进行层析过柱分离,得到3-(3-三氟甲基)-6-(2,4,6-三甲基苯氧基)哒嗪的白色固体1.4g,产率为76%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ8.34(s,1h),8.23(d,j=8hz,1h),7.93(d,j=9.2hz,1h),7.72(d,j=8.0hz,1h),7.63(t,j=8.0hz,1h),7.27(t,j=6.0hz,1h),7.27(s,2h),2.32(s,3h),2.15(s,6h);19fnmr(376mhz,cdcl3)δ-62.69(s,3f).ms((+)-esi):m/z=359.12(calcd.359.14forc20h18f3n2o,[m+h+])。
将3-(3-三氟甲基)-6-(2,4,6-三甲基苯氧基)-哒嗪1.8g(5mmol),四氯铂酸钾0.8g(2mmol),乙氧基乙醇45ml和水15ml置于120ml的封管中,在n2保护下,120℃下反应12h。反应结束后冷却至室温,用布氏漏斗进行抽虑得到黄色固体,再用二氯甲烷进行硅胶柱层析,得到配合物c的黄色固体1.1g,产率为63%。产物的核磁及质谱数据为:1hnmr(400mhz,cdcl3)δ8.22(d,j=8.4hz,2h),8.18(d,j=8.4hz,2h),7.94(s,2h),7.33-7.38(m,4h),7.28(s,4h),2.34(s,6h),2.15(s,12h);19fnmr(376mhz,cdcl3)δ-62.673(s,3f).hrms((+)-esi:m/z=910.2137(calcd.910.2155for[c40h33f6n4o2pt][m+h]+)。配合物c在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图4所示,该配合物的吸收峰位λmax=292nm,发射峰位λmax=537nm。发光量子效率为75%,5%的热失重温度为380℃。产物的结构式示意图如下所示。
实施例4
将3-(3-三氟甲基苯基)-6-氯哒嗪2.1g(8mmol)和2,2-亚甲基双(四甲基苯酚)0.9g(4mmol)置于120ml的反应瓶中,向反应瓶中加入碳酸钠1g(10mmol),将n,n-二甲基甲酰胺100ml作为溶剂,将反应瓶中通入n2密封保护,反应回流12h,反应结束冷却至室温,用乙酸乙酯进行萃取得有机相,并用水对有机相进行反萃取,有机相旋干后用石油醚和乙酸乙酯比为5:1进行层析过柱分离,得到双(5-甲基-2-((6–(3–(三氟甲基)苯基)哒嗪-3-基)氧基)苯基)甲烷的白色固体1.4g,产率为53%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ8.46(s,1h),8.37(d,j=7.6hz,1h),8.24(d,j=7.6hz,1h),8.23(s,1h),8.08(d,j=7.6hz,2h),7.67-7.76(m,8h),7.57(t,j=9.6hz,2h),7.15(s,1h),7.13(s,2h),3.86(s,2h),2.19(s,6h);19fnmr(376mhz,cdcl3)δ-62.71(s,3f),-62.74(s,3f).ms((+)-esi):m/z=673.20(calcd.673.20forc37h27f6n4o2,[m+h+])。
将双(5-甲基-2-((6-(3-(三氟甲基)苯基)哒嗪-3-基)氧基)苯基)甲烷1.7g(2.5mmol),四氯铂酸钾0.8g(2mmol),乙氧基乙醇45ml和水15ml置于120ml的封管中,在n2保护下,120℃下反应12h。反应结束后冷却至室温,用布氏漏斗进行抽虑得到黄色固体,再用二氯甲烷进行硅胶柱层析,得到配合物d的黄色固体0.7g,产率为53%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ11.53(s,2h),8.23(d,j=8.0hz,2h),8.20(d,j=8.0hz,2h),7.95(s,2h),7.35-7.36(m,4h);19fnmr(376mhz,cdcl3)δ-62.54(s,3f).hrms((+)-esi):m/z=673.2015(calcd.673.0512for[c22h12f6n4o2pt][m]+)。配合物d在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图5所示,该配合物的吸收峰位λmax=289nm,发射峰位λmax=526nm,发光量子效率为65%,5%的热失重温度为430℃。产物的结构式示意图如下所示。
实施例5
将3-三氟甲基苯硼酸1.9g(10mmol)和3,6-二氯-4,5-环己(1’,4’)基哒嗪2.3g(10mmol)置于120ml封管中,再加入碳酸钾1.4g(10mmol),pdcl2(dppf)0.2g(0.3mmol),45ml四氢呋喃和30ml水,在n2保护下加热至120℃,反应12h。反应结束冷却至室温,减压旋干四氢呋喃,再用乙酸乙酯进行萃取得有机相,有机相旋干后用石油醚和乙酸乙酯比为5:1进行层析过柱分离,得到3-氯-4,5-环己(1’,4’)基-6-(3–(三氟甲基)苯基)-哒嗪的白色固体2.3g,产率为67%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ7.91(s,1h),7.81(d,j=8.4hz,1h),7.79(d,j=9.2hz,1h),7.68(t,j=8.0hz,1h),3.56-3.59(m,1h),3.33-3.30(m,1h),2.00-1.89(m,4h),1.51-1.39(m,4h);19fnmr(376mhz,cdcl3)δ-62.68(s,3f).ms((+)-esi):m/z=339.08(calcd.339.09forc17h15clf3n2,[m+h+])。
将3-氯-4,5-环己(1’,4’)基-6-(3–(三氟甲基)苯基)-哒嗪2.7g(8mmol)和2,2-亚甲基双(四甲基苯酚)0.9g(4mmol)置于120ml的反应瓶中,向反应瓶中加入碳酸钾1g(10mmol),将n,n-二甲基甲酰胺100ml作为溶剂,将反应瓶中通入n2密封保护,反应回流12h,反应结束冷却至室温,用乙酸乙酯进行萃取得有机相,并用水对有机相进行反萃取,有机相旋干后用石油醚和乙酸乙酯比为5:1进行层析过柱分离,得到双(2-((4,5-环己(1’,4’)基-6-(3-(三氟甲基)苯基)哒嗪-3-基)氧基)-5-甲苯基)甲烷的白色固体1.4g,产率为43%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ7.89(d,j=8.4hz,2h),7.68-7.73(m,4h),7.57(d,j=7.6hz,2h),7.02(d,j=9.2hz,6h),3.85(s,2h),3.55-3.57(m,2h),3.32-3.29(m,2h),2.27(s,6h),1.82(d,j=9.6hz,8h),1.39(d,j=8.4hz,8h);19fnmr(376mhz,cdcl3)δ-62.10(s,3f),-62.51(s,3f).ms((+)-esi):m/z=833.32(calcd.833.33forc49h43f6n4o2,[m+h+])。
将双(2-((4,5-环己(1’,4’)基-6-(3-(三氟甲基)苯基)哒嗪-3-基)氧基)-5-甲苯基)甲烷2.0g(2.5mmol),四氯铂酸钾0.8g(2mmol),乙氧基乙醇45ml和水15ml置于120ml的封管中,在n2保护下,120℃下反应12个小时。反应结束后冷却至室温,用布氏漏斗进行抽虑得到黄色固体,再用二氯甲烷进行硅胶柱层析,得到配合物e的黄色固体0.9g,产率为56%。产物的核磁及质谱数据分别为:1hnmr(400mhz,cdcl3)δ11.49(s,2h),8.25(d,j=8.2hz,2h),8.19(d,j=8.2hz,2h),7.97(s,2h),4.04(s,2h),3.01(s,2h),1.84-1.96(m,8h),1.56-1.64(m,8h);19fnmr(cdcl3,376mhz)δ-61.60(s,3f),-62.68(s,3f).hrms((+)-esi):m/z=833.3283(calcd.833.1764for[c34h28f6n4o2pt][m]+)。配合物e在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图6所示,该配合物的吸收峰位λmax=289nm,发射峰位λmax=530nm。发光量子效率为80%,5%的热失重稳定为418℃。产物的结构式示意图如下所示。
- 还没有人留言评论。精彩留言会获得点赞!
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- 一种新型罗汉果醇衍生物单体的制作方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法