新型磁性微粒子聚合物的制备方法与流程
本发明涉及一种新型磁性微粒子聚合物的制备方法,用于医疗诊断、磁性导电材料、磁性功能性涂料及磁性干式粉体等领域。
背景技术:
磁性聚合物微粒子,因其具有磁性,便于简易、快速磁力回收的优点。从而被用于医疗诊断、核酸提取、蛋白分离、细菌分离、酶的精制、药物传递信号分析及相关生命科学等领域,目前被各国科研学者所重视,从目前国内文献专利cn200510087784.7制造磁性微颗粒方法,都是聚合物,外围有很多磁性微粒子,在聚合物外,通过异质凝集hetero-coagulation形成磁性聚合物微颗粒。采用这种方法有以下的缺点:
(1)微球磁含量不均匀。磁性微粒子比表面积小,磁性纳米粒子分布不均匀。聚合反应时,反应体系不稳定,得到的微球磁含量分布不均匀。
(2)磁含量不可控。由于制备壳层复合微球过程中,磁性纳米粒子通过静电吸附过程包覆在聚合物微球表面,当聚合物表面富集满磁性纳米粒子后微球表面不再继续富集磁性纳米粒子,磁含量不再继续提高。
(3)复合微球表面磁性纳米粒子容易脱落。由于磁性纳米粒子表面结合能力不强,磁性聚合物微粒子在使用时,磁性纳米粒子容易被溶出,从聚合物表面脱落下来,从而影响生物活性物质的活性,如:蛋白质、酶失活的可能,从而导致检测分析错误等现象发生。
综上所述,cn200510087784.7专利中采用上述方法聚合制成磁性微粒子在使用中受到局限性。近年来针对此种方法改良专利和文献并不多见,本发明专利对以上不足地方进行了深度改良和机理分析,避免了磁性聚合物微粒子在使用过程中容易脱落的现象和磁含量不可控以及微球表面生物相容性差等特点。
技术实现要素:
有鉴于此,本发明的目的是对以上不足进行了深度改良和机理分析,提供一种新型磁性微粒子聚合物的制备方法。
本发明解决上述现有技术的不足所采用的技术方案是:一种新型磁性微粒子聚合物的制备方法,包括如下步骤:
采用烯烃类的单体聚合合成带负电的聚合物微球,将磁性纳米粒子的分散液加入聚合物微球当中,由于合成的磁性纳米粒子表面带有正电,通过静电吸附作用在ps微球外部凝集磁性纳米粒子;
将阴离子高分子电解质与磁性纳米粒子混合均匀,将混合液加入到(1)中,通过静电吸附,形成第二次凝集反应;
将带有正电的磁性纳米粒子再次与步骤(2)溶液混合均匀,通过静电吸附,形成第三次凝集反应;
将第三次凝集后的微球加入疏水性改性剂,80℃反应30min,磁分离,水洗三次;
将步骤(4)制得的微球,水,引发剂以及乙烯类单体加入到三口烧瓶中,通n230min,升温至80℃,反应16-24h,最后得到包覆聚合物外层保护膜的磁性聚合物微粒子。
优选的,所述烯烃类的单体为苯乙烯、甲基苯乙烯、芳香族苯乙烯化合物、丙烯酸、不饱和羧酸类丙烯酸、丙烯酸、甲基苯烯酸乙酯、甲基丙烯酸丁酯、卤素乙烯类化合物。
优选的,所述磁性纳米粒子为金属离子,化合物或单质。
优选的,所述磁性聚合物微粒子粒径为0.2-60μm,所述磁性纳米粒子粒径为0.001-0.1μm,所述磁性聚合物微粒子粒径为200nm。
优选的,所述聚合物外层保护膜的保护层厚度为0.1-1μm。
优选的,所述阴离子高分子电解质为对苯乙烯磺酸钠,十二烷基硫酸钠,十二烷基苯磺酸钠其中的一种或多种组合。
优选的,所述引发剂为过硫酸钾、过硫酸钠,过硫酸铵,该引发剂的用量占烯烃类的单体的0.1%-5%。
优选的,所述疏水性改性剂为油酸钠,油酸铵,油酸,十一烯酸的一种或多种组合。
优选的,所述磁性聚合物微粒子中聚合物与磁性纳米粒子的重量比为0.5-2.6。
相较于现有技术,本发明的新型磁性微粒子聚合物的制备方法,制得粒径均一磁性聚合物微粒子的方法简单易行,具有较高的磁性和结构稳定性以及良好的生物相容性,避免了磁性聚合物微粒子在使用过程中容易脱落的现象和磁含量不可控以及微球表面生物相容性差等缺点,可以运用在生理活性物的检测、分离、提取及其它领域,实用价值广泛。
附图说明
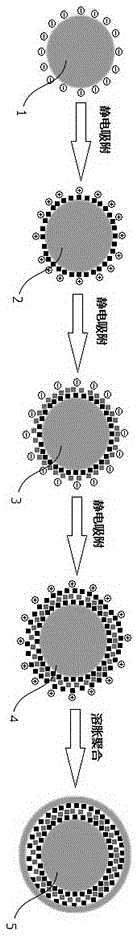
图1为本发明新型磁性微粒子聚合物的制造合成机理图。
具体实施方式
以下描述用于揭露本发明以使本领域技术人员能够实现本发明。以下描述中的优选实施例只作为举例,本领域技术人员可以想到其他显而易见的变型。在以下描述中界定的本发明的基本原理可以应用于其他实施方案、变形方案、改进方案、等同方案以及没有背离本发明的精神和范围的其他技术方案。
请参阅图1,为本发明的新型磁性微粒子聚合物的制备示意图,其包括如下步骤:
步骤(1):采用烯烃类的单体聚合合成带负电的聚合物微球1,将磁性纳米粒子的分散液加入聚合物微球当中,由于合成的磁性纳米粒子表面带有正电,通过静电吸附作用在ps微球外部凝集磁性纳米粒子形成单层磁性聚合物微粒子2;
步骤(2):将阴离子高分子电解质与磁性纳米粒子混合均匀,将混合液加入到(1)中,通过静电吸附,形成第二次凝集反应的带负电fe3o4纳米粒子凝集体形成双层磁性聚合物微粒子3;
步骤(3):将带有正电的磁性纳米粒子再次与步骤(2)溶液混合均匀,通过静电吸附,形成第三次凝集反应的带正电fe3o4纳米粒子凝集体形成多层磁性聚合物微粒子4;
步骤(4):将第三次凝集后的微球加入疏水性改性剂,80℃反应30min,磁分离,水洗三次;
步骤(5):将步骤(4)制得的微球,水,引发剂以及乙烯类单体加入到三口烧瓶中,通n230min,升温至80℃,反应16-24h,最后得到包覆聚合物外层保护膜的磁性聚合物微粒子形成磁性聚合物微粒子5。
其中,所述烯烃类的单体包括但不限于苯乙烯、甲基苯乙烯、芳香族苯乙烯化合物、丙烯酸、不饱和羧酸类丙烯酸、丙烯酸、甲基苯烯酸乙酯、甲基丙烯酸丁酯、卤素乙烯类化合物;所述磁性纳米粒子为四氧化三铁、铁、锰、钴、镍等金属离子,还包括化合物或单质;所述磁性聚合物微粒子粒径为0.2-60μm,所述磁性纳米粒子粒径为0.001-0.1μm,所述磁性聚合物微粒子粒径为200nm;所述聚合物外层保护膜的保护层厚度为0.1-1μm,保护膜粒径过大,磁性下降,运用磁性功能减弱,适用性范围受限;所述阴离子高分子电解质为对苯乙烯磺酸钠,十二烷基硫酸钠,十二烷基苯磺酸钠,其中的一种、两种或三种组合;所述引发剂为过硫酸钾、过硫酸钠,过硫酸铵,该引发剂的用量占烯烃类的单体的0.1%-5%;所述疏水性改性剂为油酸钠,油酸铵,油酸,十一烯酸,其中的一种、两种、三种或四种组合;所述磁性聚合物微粒子中聚合物与磁性纳米粒子的重量比为0.5-2.6。
以下为本发明的几个具体的实施例:
实施例1
本实施例制备一种新型磁性微粒子聚合物的制备方法:
1)向4l的三口烧瓶内注入800ml纯水和200gst,通氮气30min,升温至80℃,加入引发剂过硫酸钾2g,恒温80℃,6小时之后,st聚合完成,获得st微球,直径1.2μm,洗净后将浓度调整为10%乳液。
2)取10%乳液10ml,加入0.2g共沉淀制备的fe3o4纳米粒子0.025μm,冰水浴超声分散15min,在ps微球外部形成一层fe3o4纳米粒子异质凝集体。
3)取0.2g共沉淀制备的fe3o4,加对苯乙烯磺酸钠5g浓度为0.0025%超声15min后加入到步骤2)中。
4)取0.2g共沉淀制备的fe3o4,加入到步骤3)中,超声15min后,此时,形成多层凝集体。
5)将上述溶液离心去除上层清液,加入纯水200g,ph调整到5.0,再加入0.2%油酸钠30g,使多层异质凝集体进行表面改性,再次将溶液调整为1%的固含量。另取三口烧瓶,加st0.5g、kps0.05g,将上述1%固含量多层凝集体注入三口烧瓶中,通氮气30min,升温至70℃,恒温70℃保持6h。原本裸露的fe3o4外部被ps聚合物包被,得到粒径为1.5μm磁性聚合物微粒子。
以上方法制得磁性微粒子,经热重量分析,聚合物的fe3o4占50%,tem观察聚合物壳层厚度为0.1μm。
实施例2
本实施例制备一种新型磁性微粒子聚合物的制备方法:
1)向4l的三口烧瓶内注入800ml纯水和200gst,通氮气30min,升温至80℃,加入引发剂过硫酸胺2g,恒温80℃,6小时之后,st聚合完成,获得st微球,直径1.2μm,洗净后将浓度调整为10%乳液。
2)取10%乳液10ml,加入0.2g共沉淀制备的fe3o4纳米粒子0.025μm,冰水浴超声分散15min,在ps微球外部形成一层fe3o4纳米粒子异质凝集体。
3)取0.2g共沉淀制备的fe3o4,加十二烷基苯磺酸钠5g浓度为0.0025%超声15min后加入到步骤2)中。
4)取0.2g共沉淀制备的fe3o4,加入到步骤3)中,超声15min后,此时,形成多层凝集体。
5)将上述溶液离心去除上层清液,加入纯水200g,ph调整到5.0,再加入0.2%油酸胺30g,使多层异质凝集体进行表面改性,再次将溶液调整为1%的固含量。另取三口烧瓶,加st0.5g、kps0.05g,将上述1%固含量多层凝集体注入三口烧瓶中,通氮气30min,升温至70℃,恒温70℃保持6h,原本裸露的fe3o4外部被ps聚合物包被.得到粒径为1.5μm磁性聚合物微粒子。
以上方法制得磁性微粒子,经热重量分析,聚合物的fe3o4占50%,tem观察聚合物壳层厚度为0.1μm。
实施例3
1)纯水、单体、引发剂混合,水温70-90℃,引发剂为单体的0.1%-2%,单体与水比例10%-90%,粒径大小可以用亲水性单体用量及表面活性剂用量来控制,亲水性单体占聚合物0.1%-5%为宜,合成聚合物微球,水洗三次溶于去离子中,浓度调整至10%备用。
2)取步骤1)中得到的聚合物微球乳液10ml,加入带有正电的fe3o4纳米粒子,冰水浴超声搅拌15min,继续向溶液中加入带有负电的fe3o4纳米粒子,冰水浴超声搅拌15min,加入带有正电的fe3o4纳米粒子,冰水浴超声搅拌15min,加入油酸钠,使多层凝集从亲水性变为疏水性,疏水性便于疏水性单体的溶胀和聚合包覆,油酸钠添加量为0.1-10%。
3)向步骤2)中改性后的复合微球,加入st,kps和表面活性剂冰水浴超声溶胀30min,将溶胀后的混合液加入到装有100ml缓冲溶液的三口烧瓶中,其中缓冲溶液为ph=4.6的nh4·oh和hcl缓冲溶液,通n230min,升温至70-90℃,st在复合微球表面聚合形成ps壳层,聚合物壳层厚度可以用单体、表面活性剂用量来控制,同时反应时,ph值4-6为宜,st用量与第二步形成多层磁性粒子量1-3倍为宜。
本领域的技术人员应理解,上述描述及附图中所示的本发明的实施例只作为举例而并不限制本发明。本发明的目的已经完整并有效地实现。本发明的功能及结构原理已在实施例中展示和说明,在没有背离所述原理下,本发明的实施方式可以有任何变形或修改。
- 还没有人留言评论。精彩留言会获得点赞!