一种合成4-甲氧基巴豆酸甲酯的方法与流程
本发明属于治疗青光眼药物合成方法领域,具体涉及一种快速合成4-甲氧基巴豆酸甲酯的方法。
背景技术:
青光眼(glaucoma)是一组以视乳头萎缩及凹陷、视野缺损及视力下降为共同特征的疾病,病理性眼压增高、视神经供血不足是其发病的原发危险因素。现代医学研究已经证实,降低眼压可以有效控制青光眼病情的发展。一般来说,降低眼压主要通过以下两种途径实现,一是抑制房水的产生,二是加速房水的流出。局部使用碳酸酶抑制剂(cais),可以有效抑制房水的生成,乙酰唑胺就是一种常用的cais。但遗憾的是,乙酰唑胺长时间使用对人体损伤较大,副作用较多。为了降低药物的副作用,在乙酰唑胺的基础上,现代医学已经发展出二代的替代品,譬如多佐胺和布林唑胺。一氧化氮(no)可以有效提高房水的流出速率,从而实现对眼压的控制,硝基血管扩张剂、尼普地洛、硝普钠、sin-1和snap等均属于典型的no缓释剂。在治疗青光眼的实践中,医学工作者发现单独采用一种药物很难实现对眼压的长期准确控制,一般都需要联合用药,即cais和no缓释剂联合使用。
近年来,药物化学得到了长足的发展,药物的分子结构设计更趋合理,先进的合成手段使得设计的新型药物分子的制备成为了可能。对于治疗青光眼的药物而言,如果能设计出一个新的化合物,这个化合物具备cais的母体结构,同时还可以缓慢释放no,那么可以更好的实现对眼压的准确控制。
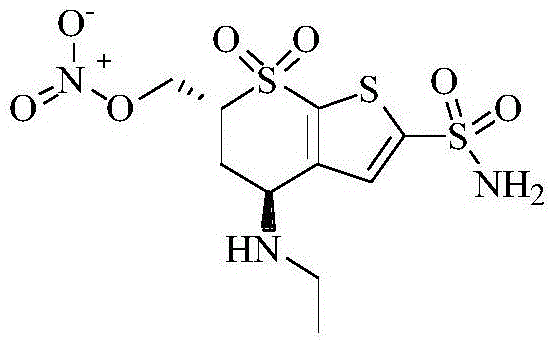
基于该理念,pfizer公司(wo2009/007814a1)发明了一种新的控制眼压的药物no-cais,其分子结构如图1所示。no-cais的母体结构为多佐胺,与多佐胺不同的是,在多佐胺的6位上引入了-ono2基团。动物实验表明,no-cais供体可以在兔眼内缓慢释放出no,进而加快了房水的流出,实现了对眼压的有效控制。该类药物有望在临床上得到应用。
4-甲氧基巴豆酸甲酯是合成no-cais的主要原料之一,它具有α、β-活泼不饱和羰基结构,可以与2-巯基噻吩反应生成no-cais的母体环状结构,在该母体结构上进一步进行功能团的修饰,即可制得no-cais,具体的合成路线如图2所示。
4-溴巴豆酸甲酯和甲醇在催化剂的作用下即可生成4-甲氧基巴豆酸甲酯。催化剂可以加速反应的进行,促进原料的转化。fizer公司采用ag2o作为催化剂,在超声反应器内合成了4-甲氧基巴豆酸甲酯。ag2o属于贵金属催化剂,成本较高,另外该反应时间较长,至少需要12-16h,生产效率较低。americancyanamidcompany公布了另一种合成4-甲氧基巴豆酸甲酯的方法(us6251912b1)。4-溴巴豆酸甲酯、ca2co3和甲醇回流反应5天,即可制得4-甲氧基巴豆酸甲酯。与ag2o相比,ca2co3易得且成本低廉,适合在工业上大规模使用。不过遗憾的是,该反应需要的时间更长。重复该实验,我们发现,反应进行5天,原料4-溴巴豆酸甲酯仍有接近2%的残留,残留的原料会给产品的提纯增加难度,这是因为原料与产品的沸点非常接近,采用常规的蒸馏方法很难将未反应的原料除去,这会造成产品中单杂过高,而且还会导致合成收率的降低。
技术实现要素:
发明目的:缩短合成4-甲氧基巴豆酸甲酯的反应时间,提高原料的转化率,简化后处理操作,降低生产成本。
本发明提供了一种高效合成4-甲氧基巴豆酸甲酯的工艺。具体操作如下:将4-溴巴豆酸甲酯、甲醇和催化剂加入至密封的反应器内,加热至65℃以上反应,原料反应完全后,将反应液降温,然后过滤,除去固体副产物,滤液浓缩,回收过量的甲醇,获得的浓缩液粗品减压蒸馏提纯,即可获得高纯度的4-甲氧基巴豆酸甲酯。
本发明所述的密封的反应器包括高压反应釜、水热反应釜、固定床反应器、喷射反应器以及管式反应器等,优选的密封反应器为高压反应釜。
本发明所述的催化剂包括碳酸钙、碳酸镁、碳酸钠和碳酸钾,催化剂可以单独使用,也可混合使用。
本发明所述的催化剂与4-溴巴豆酸甲酯的摩尔比为0.5:1-20:1,优选的催化剂与4-溴巴豆酸甲酯的摩尔比为1:1-1.5:1。
以4-溴巴豆酸甲酯计,本发明所述的甲醇用量为0.3-4l/mol,优选的甲醇用量为0.6-1.5l/mol。
本发明所述的反应温度为65-200℃,优选的反应温度为100-120℃。
本发明所述的反应时间为1-20h,优选的反应时间为2-3h。
本发明所述的反应压力为0.1-0.8mpa,优选的反应压力为0.4-0.5mpa。
本发明的优点在于:
1、采用本发明提供的工艺合成4-甲氧基巴豆酸甲酯,可以将反应时间从5天缩短至2-3h,极大的提高了合成效率;
2、本发明采用的催化剂价格低廉,生产的成本较低,适合在工业上规模化应用;
3、采用本发明提供的工艺合成4-甲氧基巴豆酸甲酯,实现了原料4-溴巴豆酸甲酯的完全转化,简化了后处理操作,提高了反应收率,显著降低了生产成本;
4、本发明虽然在合成时具有一定的压力,但是因为压力较低,故操作的安全性较高。
附图说明:
图1、no-cais分子结构式;
图2、no-cais合成路线;
图3、4-甲氧基巴豆酸甲酯的1hnmr谱图。
具体实施方式
以下结合具体实施例对上述方案做进一步说明。需要着重强调的是,这些实施例是用于说明本发明而不是限制本发明的适用范围。
实施例1:
打开100l高压反应釜的加料阀门,依次向其中加入6kg4-溴巴豆酸甲酯、5.03kg碳酸钙和50l甲醇,然后关闭加料阀门,打开氮气阀门,向反应釜内充入氮气,当压力升至0.3-0.4mpa时,关闭氮气阀门,保压10min,然后将氮气放空。氮气放空后,再向反应釜内充入氮气,维持压力在0.3-0.4mpa之间,继续保压10min,再放空。该操作再重复1次。氮气置换完成后,开启加热,反应液缓慢升温至100-120℃,此时反应釜内压力为0.4-0.5mpa,在该温度和压力下继续反应2-3h。反应采用气相色谱监控,原料4-溴巴豆酸甲酯反应完全后,关闭加热,向反应釜夹套内通入冷却水,当反应液温度降温至室温后,打开放料阀门,将反应液放出。抽滤,滤饼用少量的甲醇洗涤1-2次,合并滤液,减压浓缩,回收过量的甲醇。减压浓缩获得的粗品再经减压蒸馏提纯,即可获得4-甲氧基巴豆酸甲酯,纯度:99.5area%,单杂<0.1area%,收率98%。
实施例2:
打开100l高压反应釜的加料阀门,依次向其中加入12kg4-溴巴豆酸甲酯、7.1kg碳酸钠和40l甲醇,然后关闭加料阀门,打开氮气阀门,向反应釜内充入氮气,当压力升至0.3-0.4mpa时,关闭氮气阀门,保压10min,然后将氮气放空。氮气放空后,再向反应釜内充入氮气,维持压力在0.3-0.4mpa之间,继续保压10min,再放空。该操作再重复1次。氮气置换完成后,开启加热,反应液缓慢升温至190-200℃,此时反应釜内压力为0.7-0.8mpa,在该温度和压力下继续反应1h。反应采用气相色谱监控,原料4-溴巴豆酸甲酯反应完全后,关闭加热,向反应釜夹套内通入冷却水,当反应液温度降温至室温后,打开放料阀门,将反应液放出。抽滤,滤饼用少量的甲醇洗涤1-2次,合并滤液,减压浓缩,回收过量的甲醇。减压浓缩获得的粗品再经减压蒸馏提纯,即可获得4-甲氧基巴豆酸甲酯,纯度:99.5area%,单杂<0.1area%,收率96%。
实施例3:
打开100l高压反应釜的加料阀门,依次向其中加入2kg4-溴巴豆酸甲酯、18.8kg碳酸镁和45l甲醇,然后关闭加料阀门,打开氮气阀门,向反应釜内充入氮气,当压力升至0.3-0.4mpa时,关闭氮气阀门,保压10min,然后将氮气放空。氮气放空后,再向反应釜内充入氮气,维持压力在0.3-0.4mpa之间,继续保压10min,再放空。该操作再重复1次。氮气置换完成后,开启加热,反应液缓慢升温至65℃,此时反应釜内压力为0.1-0.2mpa,在该温度和压力下继续反应20h。反应采用气相色谱监控,原料4-溴巴豆酸甲酯反应完全后,关闭加热,向反应釜夹套内通入冷却水,当反应液温度降温至室温后,打开放料阀门,将反应液放出。抽滤,滤饼用少量的甲醇洗涤1-2次,合并滤液,减压浓缩,回收过量的甲醇。减压浓缩获得的粗品再经减压蒸馏提纯,即可获得4-甲氧基巴豆酸甲酯,纯度:99.6area%,单杂<0.1area%,收率97%。
实施例4:
打开100l高压反应釜的加料阀门,依次向其中加入6kg4-溴巴豆酸甲酯、2.32kg碳酸钾和10l甲醇,然后关闭加料阀门,打开氮气阀门,向反应釜内充入氮气,当压力升至0.3-0.4mpa时,关闭氮气阀门,保压10min,然后将氮气放空。氮气放空后,再向反应釜内充入氮气,维持压力在0.3-0.4mpa之间,继续保压10min,再放空。该操作再重复1次。氮气置换完成后,开启加热,反应液缓慢升温至150-160℃,此时反应釜内压力为0.6-0.7mpa,在该温度和压力下继续反应10h。反应采用气相色谱监控,原料4-溴巴豆酸甲酯反应完全后,关闭加热,向反应釜夹套内通入冷却水,当反应液温度降温至室温后,打开放料阀门,将反应液放出。抽滤,滤饼用少量的甲醇洗涤1-2次,合并滤液,减压浓缩,回收过量的甲醇。减压浓缩获得的粗品再经减压蒸馏提纯,即可获得4-甲氧基巴豆酸甲酯,纯度:99.8area%,单杂<0.1area%,收率94%。
上述实施例获得的4-甲氧基巴豆酸甲酯采用核磁来确认结构,其代表性的核磁谱图如图3所示。
需要重点说明的是,上述实例只是为了说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人能够了解本发明的内容并据此实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!