一种鉴定驴源性成分的特异性引物、试剂盒及鉴别方法与流程
本发明属于中药材鉴定技术领域,具体涉及一种鉴定驴源性成分的特异性引物、试剂盒及鉴别方法。
背景技术:
阿胶(asinicoriicolla)为马科动物驴(equusasinml.)的干燥皮或鲜皮经煎煮、浓缩,并可加入适量的黄酒、冰糖及豆油制成的固体胶,具有补血滋阴,润燥,止血的功效,用于治疗血虚萎黄、眩晕心悸、肌痿无力、心烦不眠等症状。阿胶为《中国药典》2015版的收录品种,其药用历史悠久,始载于《神农本草经》,被列为“上品”,称其“可久服轻身益气”,是药食同源的重要物种,尤其在治未病方面功效显著。关于阿胶的医方医案有3200多个,其中包括200多个膏方和200多个食疗方。现代研究表明阿胶能调节身体机能,增强免疫力,在肿瘤辅助治疗、血液病等重大疾病防治上,其治疗优势也正逐步凸显。我国药典明确规定生产阿胶的原料是驴皮。然而,我国驴养殖业发展严重滞后,驴的存栏数呈逐年下滑之势。以致近年制胶原料驴皮短缺、原料价格上涨,造成目前阿胶市场混乱,掺杂使假情况屡屡发生,主要掺假成分包括猪皮、牛皮胶、马皮胶甚至皮革下脚料等,给大众用药安全带来重大隐患,因此,建立有效的阿胶质量控制方法是当前急需解决的问题。
传统的阿胶真伪鉴别方法,采用通过产品的外观、颜色、气味等实现,需要极为丰富的个人经验和深厚的专业知识,而各种杂皮胶的制备工艺与正品相似,故其外观性状极为相近,更大大增加了鉴别的难度。
中国专利文献cn105586420a中公开了一种准确鉴别阿胶原料驴源性成分的特异性引物对及其方法,通过dna提取、pcr特异性扩增、酶切、电泳检测可以鉴别阿胶原料即驴皮中是否存在驴源性成分。中国专利文献cn106636385a公开了一种用于驴源性的实时荧光pcr检测的特异性引物和探针,通过dna提取、pcr特异性扩增、荧光检测可以鉴别肉食原料是否存在驴源性成分。以上两个专利均是对驴的直接产品(驴皮或驴肉)进行鉴定,难度相对较低,直接提取即可得到高质量dna,然而阿胶为深加工产品,在阿胶的生产过程中,驴皮经高温长时间煎煮,dna遭到严重破坏,成品中dna含量少而且片段极短,而且,阿胶中主要成分为各种胶原蛋白肽段,大量蛋白的存在会严重影响dna提取的效率,此外,制备阿胶成品的过程中,不仅要经过长时间煎煮,而且后期要加入黄酒、豆油等辅料,这些辅料的加入会加大提取成品阿胶dna的难度并引入多种抑制pcr反应的成分,以上原因使得阿胶中驴源性成分的鉴别难度很大,使用上述现有技术的方法鉴定阿胶中驴源性成分时,易出现扩增不出来,杂带等问题,准确度、灵敏度低,特异性差,均不适用于对阿胶的鉴别。
技术实现要素:
本发明要解决的第一个技术问题在于提供一种鉴定驴源性成分的特异性引物,采用所述引物鉴定驴源性成分,鉴定结果准确、稳定、可靠、灵敏度高,能够在短时间内对大量待测混合物中驴源性成分进行快速鉴定,效率高。
本发明要解决的第二个技术问题在于提供一种鉴定驴源性成分的试剂盒,采用所述试剂盒鉴定驴源性成分,鉴定结果准确、稳定、可靠、灵敏度高,能够在短时间内对大量待测混合物中驴源性成分进行快速鉴定,效率高。
本发明要解决的第三个技术问题在于提供一种鉴定驴源性成分的方法,采用所述方法鉴定驴源性成分,鉴定结果准确、稳定、可靠、灵敏度高,能够在短时间内对大量待测混合物中驴源性成分进行快速鉴定,效率高。
为此,本发明提供了一种用于鉴定驴源性成分的引物,所述引物如下:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
本发明提供了一种鉴定驴源性成分的试剂盒,含有上述的引物。
所述的试剂盒,每支所述试剂盒包括各自独立包装的引物液部分和pcr反应液部分;所述pcr反应液部分含有用于进行pcr扩增的扩增缓冲液、dntps、taqdna聚合酶以及无菌超纯水。
本发明还提供了一种所述的引物或所述的试剂盒在鉴定驴源性成分中的应用;
进一步地,所述混合物为驴皮、阿胶半成品、阿胶成品、含阿胶的药物组合物和含阿胶的药物制剂中的至少一种。
本发明提供了一种鉴定驴源性成分的方法,包括如下步骤:
(1)待测样品进行预处理,得预处理样品;
(2)提取预处理样品中驴源性成分的总dna,备用;
(3)以步骤(2)中提取的dna为模板,采用如下引物进行pcr扩增:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′;
(4)测定步骤(3)中所得的pcr扩增产物的大小,若所述pcr扩增产物中含有50~150bp的dna片段,则所述待测样品存在驴源性成分;反之,则所述待测样品不存在驴源性成分。
优选地,所述待测样品预处理方法为ni-nta预处理;
所述ni-nta预处理的具体步骤为:取待测样品10~100重量份,加1~10体积份平衡缓冲液,温浴烊化至样品融化完全,置于ni-nta预装柱内,于4℃下震摇结合0.5~2h,收集穿透液,80~100℃蒸干,即得预处理样品;所述重量份与体积份的比值关系为mg/ml。
所述步骤(3)中,所述pcr反应体系包括:
dna模板,50ng/μl,1~5μl;
上游引物,10μm,0.5~1.5μl;
下游引物,10μm,0.5~1.5μl;
taqdna聚合酶,5u/μl,0.2~1.2μl;
dntps,10mm,0.25~1μl;
10×pcr扩增缓冲液,1.5~4.5μl;
无菌超纯水补至25μl;
优选的,所述pcr扩增程序如下:95℃预变性5min;94℃变性30s,48~68℃退火30s,72℃延伸30s,进行30~40个循环;然后继续72℃延伸5~10min,降温至4℃,获得的扩增产物4℃保存。
所述pcr扩增程序中,退火温度为58℃。
所述步骤(4)中所述50~150bp的dna片段为101bp的dna片段。
所述的101bp的dna片段序列如seqidno:7所示。
本发明技术方案,具有如下优点:
(1)本发明所述的用于鉴定驴源性成分的特异性引物,基于驴的cytb和cox-1基因设计了大量的引物,且通过大量引物筛选研究,筛选出了具有种属特异性的引物,使用上述引物对不仅能够用于简单加工的驴皮、长时间煮沸得到的阿胶半成品,还能够对深加工的阿胶产品以及含有阿胶产品的药物组合物和药物制剂中驴源性成分进行鉴定,鉴定结果准确、稳定、可靠、灵敏度高,并能将驴源性成分与其他亲源关系比较近的种属混淆品准确的分开,特异性高,且其pcr扩增产物仅为50~150bp,分子量小,保证了其在短时间内进行大量的阿胶中驴源性成分鉴定。
(2)本发明所述的用于鉴定驴源性成分的试剂盒,所述试剂盒中含有所述的用于鉴定驴源性成分的特异性引物,所述引物具有种属特异性,使用上述引物对待测混合物中驴源性成分进行鉴定,能将驴源性成分与其他亲源关系比较近的种属混淆品准确的分开,在短时间内进行大量的混合物中驴源性成分鉴定,鉴定结果准确、稳定、可靠、灵敏度高。
(3)本发明所述的鉴定驴源性成分的方法,由于阿胶为深加工产品,在生产过程中存在较长高温煎煮及浓缩过程,使dna严重降解;且阿胶主要成分为蛋白质成分,在dna提取过程中存在污染,易影响后续扩增实验,本发明的鉴定方法首先通过对待测阿胶用ni-nta预处理,得预处理样品,明显提高dna样品的纯度,且对dna破坏性低,然后提取预处理样品中驴源性成分的总dna,以提取的dna为模板,采用50~150bp的dna片段为目的片段的引物进行pcr扩增,保证了其在短时间内进行大量的混合物中驴源性成分鉴定,鉴定结果准确、稳定、可靠、灵敏度高。
(4)本发明所述的鉴定驴源性成分的方法,所述步骤(3)中,所述50~150bp的dna片段为101bp的dna片段,pcr扩增产物分子量小,鉴定结果准确、稳定、可靠、灵敏度高。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单的介绍,很显然下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为采用不同试剂盒鉴定驴源性成分的琼脂糖凝胶电泳图;
图2为以101bp的dna为的目的片段得到的琼脂糖凝胶电泳图;
图3为以155bp的dna为的目的片段得到的琼脂糖凝胶电泳图;
图4为以211bp的dna为的目的片段得到的琼脂糖凝胶电泳图;
图5为不同种属样品的琼脂糖凝胶电泳图,其中;
图6为实施例3、实施例4、对比例5和对比例6中的琼脂糖凝胶电泳图。
具体实施方式
本发明所使用的主要样品和试剂如下:
阿胶样品1,厂家:北京同仁堂,批号:15191758;
阿胶样品2,厂家:北京同仁堂,批号:15191761;
阿胶样品3,厂家:北京同仁堂,批号:15191768;
牛皮胶:将牛皮浸泡去毛,切块洗净,分次水煎,滤过,合并滤液,浓缩至稠膏状,冷凝即可;
猪皮胶:将猪皮浸泡去毛,切块洗净,分次水煎,滤过,合并滤液,浓缩至稠膏状,冷凝即可;
马皮胶:将马皮浸泡去毛,切块洗净,分次水煎,滤过,合并滤液,浓缩至稠膏状,冷凝即可;
空白对照:纯水或不加任何物质;
takaraminibestdna试剂盒(宝日医生物技术有限公司)和gmofooddnaextractionkitpcr深加工食品dna提取试剂盒(天根生化科技(北京)有限公司)。
本发明所使用的主要设备如下:
5424r高速冷冻离心机购自艾本德,mastercycleprospcr仪购自艾本德,bio-radpowerpacbasic电泳仪购自伯乐,bio-radmolecularimagegeldoctmxr+凝胶成像系统购自伯乐。
一、试剂盒的筛选实验
(1)待测阿胶用ni-nta预处理
取阿胶样品1、阿胶样品2和阿胶样品3,分别按照如下步骤,进行预处理,得到预处理样品1、预处理样品2和预处理样品3。
取50mg阿胶样品,加5mllysisbuffer(nah2po4浓度为50mm(mm为mmol/l),nacl浓度为300mm,naoh调节ph至8.0),温浴烊化至样品融化完全,置于ni-nta预装柱内(填料量按照每1mlni-nta填料结合10mg蛋白计),于冰上震摇结合1h,收集穿透液,85℃蒸干,即得预处理样品。
(2)dna提取
取预处理样品1、预处理样品2和预处理样品3分别采用takaraminibestdna试剂盒和gmofooddna试剂盒提取总dna,按照说明书操作,步骤如下:
①takaraminibestdna试剂盒
1、取2~25mg的动物组织、深加工品或25~100mg植物组织(切勿使用超过最大起始量的组织),置于2ml离心管中,用剪刀尽可能地剪成碎块,对于一些质地坚硬的组织也可以进行液氮研磨。
2、加入180μl的buffergl、20μl的proteinasek和10μl的rnasea(10mg/ml),于56℃水浴温浴至组织完全裂解(2~3小时,难裂解的材料可以适当延长裂解时间甚至过夜裂解。植物材料可能残存纤维状组织无法完全裂解,裂解之后先12000rpm离心2分钟,去除杂质之后再进行后续操作)。
3、向裂解液中加入200μlbuffergb和200μl100%乙醇,充分吸打混匀。
4、将spincolumn安置于collectiontube上,溶液移至spincolumn中,12000rpm离心2分钟,弃滤液。
5、将500μl的bufferwa加入至spincolumn中,12000rpm离心1分钟,弃滤液。
6、将700μl的bufferwb加入至spincolumn中,12000rpm离心1分钟,弃滤液。
注)请确认bufferwb中已经加入指定体积的100%乙醇。
7、重复操作步骤6.
8、将spincolumn安置于collectiontube上,12000rpm离心2分钟。
9、将spincolumn安置于新的1.5ml的离心管上,在spincolumn膜的中央处加入50~200μl的灭菌蒸馏水或elutionbuffer,室温静置5分钟。注)将灭菌蒸馏水或elutionbuffer加热至65℃使用时有利于提高洗脱效率。
10、12000rpm离心2分钟洗脱dna。
如需获得更大收量,可将离下液重新加入到spincolumn膜的中央或再加入50~200μl的灭菌水或elutionbuffer,室温静置5分钟后,12000rpm离心2分钟洗脱dna。
11、基因组dna定量。
提取得到的基因组dna可通过电泳或吸光度测定以定量。
②gmofooddna试剂盒
1、称取研碎的深加工食品100mg或预处理样品,加500μl缓冲液gmo1和20ul的proteinasek(20mg/ml),漩涡震荡1min。
2、56℃孵育1h。孵育过程每15min震荡一次。
3、加入200μl缓冲液gmo2.充分混匀,漩涡震荡1min。室温静置10min。
4、12000rpm(~13400×g)离心5min,将上清转移至新的离心管中。
5、向上请液中加入0.7倍体积的异丙醇,充分混匀。(例如500μl的水相溶液加350μl异丙醇),12000rpm(~13400×g)离心3min,弃上清,保留沉淀(此步沉淀可能看不见)。
注意:加异丙醇沉淀后,弃上清应小心,防止把dna倒出。
6、可选步骤:在步骤5加异丙醇之前,向上清液中加入1μlcarrierrna(酱油、番茄酱必加),再加入异丙醇。
7、加入700μl70%乙醇,漩涡震荡5sec,12000rpm(~13400×g)离心2min,弃上清。
8、重复操作步骤7.
注意:弃上清应小心,防止把dna倒出。
9、开盖倒置,室温5-10min,彻底晾干残余的乙醇。
注意:乙醇的残留会影响后续的酶反应(pcr、荧光定量pcr等)实验。
10、加入20-50μl洗脱液te,漩涡振荡1min,最终得到dna溶液。
(3)pcr扩增
以步骤(2)中提取的dna为模板,采用如下引物进行pcr扩增:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
所述pcr反应体系包括:
dna模板,50ng/μl,3μl;
上游引物,10μm,1μl;
下游引物,10μm,1μl;
taqdna聚合酶,5u/μl,0.4μl;
dntps,10mm,0.5μl;
10×pcr扩增缓冲液,2.5μl;
无菌超纯水补至25μl。
扩增反应在pcr仪上进行,所述pcr扩增程序如下:
95℃预变性5min;95℃变性30s,58℃退火30s,72℃延伸30s,进行40个循环;然后继续72℃延伸7min,降温至4℃。
(4)凝胶电泳
取步骤(3)中的pcr扩增产物分别进行琼脂糖凝胶电泳,测得各样品的pcr扩增产物的大小,琼脂糖凝胶电泳法按照2015年版中国药典三部附录ⅳb,胶浓度为2%,核酸燃料gelred浓度为1%,电泳缓冲液为tae缓冲液(tris-edta-乙酸缓冲液,ph8.3),上述步骤(2)中获得的各样品的pcr反应溶液的上样量分别为10μl,dna分子量标记(dnamarker)上样量为5μl,电泳结束后,在凝胶成像仪上观察结果。若所述pcr扩增产物中含有101bp的dna片段,则所述待测阿胶存在驴源性成分;反之,则所述待测阿胶不存在驴源性成分。
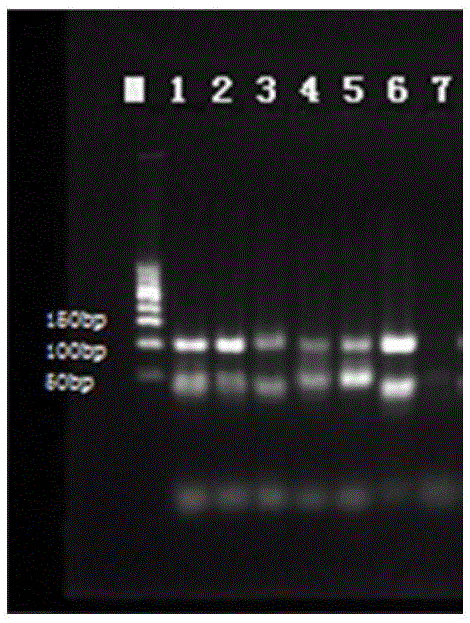
结果见图1所示,其中m为dnamarker(50bp),条带1-3分别为待测样品1、待测样品2和待测样品3采用takaraminibestdna试剂盒提取dna样品pcr产物;条带4-6分别为待测样品1、待测样品2和待测样品3采用gmofooddna试剂盒提取dna样品pcr产物;从图中可以看出,不同厂家的试剂盒提取的待测样品1、待测样品2和待测样品3的dna的扩增结果无明显差异,均能在101bp处显现明显的条带,检测效果良好,方法适用性高。
二、引物的筛选实验
(1)待测阿胶用ni-nta预处理
取阿胶样品1和阿胶样品2,分别按照如下步骤,进行预处理,得到预处理样品1和预处理样品2。
取50mg阿胶样品,加5mllysisbuffer(nah2po450mm,nacl300mm,naoh调节ph至8.0),温浴烊化至样品融化完全,置于ni-nta预装柱内(填料量按照每1mlni-nta填料结合10mg蛋白计),于冰上震摇结合1h,收集穿透液,85℃蒸干,即得预处理样品。
(2)dna提取
取预处理样品1和预处理样品2分别采用takaraminibestdna试剂盒提取总dna,按照说明书操作,且与前述takaraminibestdna试剂盒的提取步骤相同。
(3)pcr扩增
为得到目的片段为101bp、155bp和211bp的扩增产物,以步骤(2)中提取的dna为模板,分别采用如下引物对进行pcr扩增:
第一引物对如下,参见序列表中seqidno:1-2所示:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
所得扩增片段序列如下,参见序列表中seqidno:7所示:
aggagcaacggtcattacaaacctcctatcagcaatcccctacatcggtactacactcgtcgaatggatctgaggtggattctcagtagacaaagccaccc。
第二引物对如下,参见序列表中seqidno:3-4所示:
上游引物:5′-cccttagtcaatgcgtgtt-3′;
下游引物:5′-gcttgcgagtggtatgaag-3′。
所得扩增片段序列如下,参见序列表中seqidno:8所示:
cccttagtcaatgcgtgttctgactcttagtagcagacttactaacattaacatgaatcggtggccaaccagtagaacacccatacgtaatcatcggccaactggcctcaatcctctacttctccctaattctcatcttcataccactcgcaagc。
第三引物对如下,参见序列表中seqidno:5-6所示
上游引物:5′-atcatcacagccctggtaa-3′;
下游引物:5′-gggtgtagttgtctgggtct-3′。
所得扩增片段序列如下,参见序列表中seqidno:9所示:
atcatcacagccctggtaatcgtccatctactattcctccacgaaacaggatccaacaacccctcaggaatcccatctgacatagacaaaatcccattccacccgtactacacaattaaagacatcctaggacttcttctcctagtcctactcctactaaccctagtattattctcccctgacctcctaggagacccagacaactacaccc。
所述pcr反应体系包括:
dna模板,50ng/μl,3μl;
上游引物,10μm,1μl;
下游引物,10μm,1μl;
taqdna聚合酶,5u/μl,0.4μl;
dntps,10mm,0.5μl;
10×pcr扩增缓冲液,2.5μl;
无菌超纯水补至25μl。
扩增反应在pcr仪上进行,所述pcr扩增程序如下:
95℃预变性5min;95℃变性30s,58℃退火30s,72℃延伸30s,进行40个循环;然后继续72℃延伸7min,降温至4℃。
(4)凝胶电泳
取步骤(3)中的pcr扩增产物分别进行琼脂糖凝胶电泳,测得各样品的pcr扩增产物的大小,琼脂糖凝胶电泳法按照2015年版中国药典三部附录ⅳb,胶浓度为2%,核酸燃料gelred浓度为1%,电泳缓冲液为tae缓冲液(tris-edta-乙酸缓冲液,ph8.3),上述步骤(2)中获得的各样品的pcr反应溶液的上样量分别为10μl,dna分子量标记(dnamarker)上样量为5μl,电泳结束后,在凝胶成像仪上观察结果。结果见图2-4中所示。
如图2所示,目的片段为101bp的dna时,m为dnamarker(50bp),条带1为阿胶样品1,条带2为阿胶样品2,条带3为空白对照,从图中可见,阿胶样品的dna可以扩增为较为清晰的目的条带,如图3所示,目的片段为155bp的dna时,m为dnamarker(50bp),条带1为阿胶样品1的pcr产物,条带2为阿胶样品2的pcr产物,条带3为空白对照,从图中看出,阿胶样品的dna不能有效扩增,杂带较多,如图4所示,目的片段为211bp的dna,m为dnamarker(50bp),条带1为阿胶样品1,条带2为阿胶样品2,条带3为空白对照,从图中可知,阿胶样品中无有效片段扩增。
三、特异性考察实验
为保证分子鉴别结果的可靠性,本实验收集了牛皮胶:猪皮胶、马皮胶为待测样品,以对本实验方法进行适用性验证,确认本方法准确、可靠。
(1)待测样品用ni-nta预处理
分别取50mg待测样品,加5mllysisbuffer(nah2po450mm,nacl300mm,naoh调节ph至8.0),温浴烊化至样品融化完全,置于ni-nta预装柱内(填料量按照每1mlni-nta填料结合10mg蛋白计),于冰上震摇结合1h,收集穿透液,85℃蒸干,即得预处理样品。
(2)dna提取
取预处理样品采用takaraminibestdna试剂盒提取总dna,按照说明书操作,且与前述takaraminibestdna试剂盒的提取步骤相同。
(3)pcr扩增
以步骤(2)中提取的dna为模板,采用如下引物进行pcr扩增:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
所述pcr反应体系包括:
dna模板,50ng/μl,3μl;
上游引物,10μm,1μl;
下游引物,10μm,1μl;
taqdna聚合酶,5u/μl,0.4μl;
dntps,10mm,0.5μl;
10×pcr扩增缓冲液,2.5μl;
无菌超纯水补至25μl。
扩增反应在pcr仪上进行,所述pcr扩增程序如下:
95℃预变性5min;95℃变性30s,58℃退火30s,72℃延伸30s,进行40个循环;然后继续72℃延伸7min,降温至4℃。
(4)取步骤(3)中的pcr扩增产物进行琼脂糖凝胶电泳,测得各样品的pcr扩增产物的大小,琼脂糖凝胶电泳法按照2015年版中国药典三部附录ⅳb,胶浓度为2%,核酸燃料gelred浓度为1%,电泳缓冲液为tae缓冲液(tris-edta-乙酸缓冲液,ph8.3),上述步骤(2)中获得的各样品的pcr反应溶液的上样量分别为10μl,dna分子量标记(dnamarker)上样量为5μl,电泳结束后,在凝胶成像仪上观察结果,结果如图5所示,m为dnamarker(50bp),条带1为牛皮胶,条带2为猪皮胶,条带3为马皮胶,条带4为阿胶,条带5为空白对照,只有条带4对应的阿胶能够扩增得到目的片段,而与驴皮种属接近的猪皮胶、牛皮胶、马皮胶均不能扩增得到目的片段,无杂带,说明本发明鉴定方法具有优良的种属特异性。
实施例1引物
本实施例提供了一种用于鉴定驴源性成分的特异性引物,针驴的cytb基因设计的特异性引物如下,参见序列表中seqidno:1-2所示:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
上述引物由生工生物工程(上海)股份有限公司合成。
实施例2试剂盒
本实施例提供了一种用于鉴定驴源性成分的试剂盒,每支所述试剂盒包括各自独立包装的引物液部分和pcr反应液部分;所述引物液为含所述实施例1中的所述特异性引物;所述pcr反应液部分含有用于进行pcr扩增的扩增缓冲液、dntps、taqdna聚合酶以及无菌超纯水,每支所述试剂盒的配方为:
dna模板,50ng/μl,3μl;
上游引物,10μm,1μl;
下游引物,10μm,1μl;
taqdna聚合酶,5u/μl,0.4μl;
dntps,10mm,0.5μl;
10×pcr扩增缓冲液,2.5μl;
无菌超纯水补至25μl。
实施例3鉴定方法
本实施例提供了一种利用实施例1的特异性引物或实施例2的试剂盒鉴定驴源性成分的方法,包括如下步骤:
(1)待测阿胶用ni-nta预处理
取50mg阿胶样品1,加5mllysisbuffer(nah2po450mm,nacl300mm,naoh调节ph至8.0),温浴烊化至样品融化完全,置于ni-nta预装柱内(填料量按照每1mlni-nta填料结合10mg蛋白计),于冰上震摇结合1h,收集穿透液,85℃蒸干,即得预处理样品。
(2)dna提取
取预处理样品采用takaraminibestdna试剂盒提取总dna,按照说明书操作,且与前述takaraminibestdna试剂盒的提取步骤相同。
(3)pcr扩增
以步骤(2)中提取的dna为模板,采用如下引物进行pcr扩增:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
所述pcr反应体系包括:
dna模板,50ng/μl,3μl;
上游引物,10μm,1μl;
下游引物,10μm,1μl;
taqdna聚合酶,5u/μl,0.4μl;
dntps,10mm,0.5μl;
10×pcr扩增缓冲液,2.5μl;
无菌超纯水补至25μl。
扩增反应在pcr仪上进行,所述pcr扩增程序如下:
95℃预变性5min;95℃变性30s,58℃退火30s,72℃延伸30s,进行40个循环;然后继续72℃延伸7min,降温至4℃。
(4)取步骤(3)中的pcr扩增产物进行琼脂糖凝胶电泳,测得各样品的pcr扩增产物的大小,琼脂糖凝胶电泳法按照2015年版中国药典三部附录ⅳb,胶浓度为2%,核酸燃料gelred浓度为1%,电泳缓冲液为tae缓冲液(tris-edta-乙酸缓冲液,ph8.3),上述步骤(2)中获得的各样品的pcr反应溶液的上样量分别为10μl,dna分子量标记(dnamarker)上样量为5μl,电泳结束后,在凝胶成像仪上观察结果。若所述pcr扩增产物中含有101bp的dna片段,则所述待测阿胶存在驴源性成分;反之,则所述待测阿胶不存在驴源性成分。
实施例4鉴定方法
本实施例提供了一种利用实施例1的特异性引物或实施例2的试剂盒鉴定驴源性成分的方法,包括如下步骤:
(1)待测阿胶用ni-nta预处理
取100mg阿胶样品2,加1mllysisbuffer(nah2po475mm,nacl100mm,naoh调节ph至10.0),温浴烊化至样品融化完全,置于ni-nta预装柱内(填料量按照每1mlni-nta填料结合8mg蛋白计),于冰上震摇结合2h,收集穿透液,80℃蒸干,即得预处理样品。
(2)dna提取
取预处理样品采用takaraminibestdna试剂盒提取总dna,按照说明书操作,且与前述takaraminibestdna试剂盒的提取步骤相同。
(3)pcr扩增
以步骤(2)中提取的dna为模板,采用如下引物进行pcr扩增:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
所述pcr反应体系包括:
dna模板,50ng/μl,5μl;
上游引物,10μm,0.5μl;
下游引物,10μm,1.5μl;
taqdna聚合酶,5u/μl,0.2μl;
dntps,10mm,1μl;
10×pcr扩增缓冲液,1.5μl;
无菌超纯水补至25μl。
扩增反应在pcr仪上进行,所述pcr扩增程序如下:
95℃预变性5min,95℃变性30s,68℃退火30s,72℃延伸30s,进行30个循环,然后继续72℃延伸10min,降温至4℃。
(4)取步骤(3)中的pcr扩增产物进行琼脂糖凝胶电泳,测得各样品的pcr扩增产物的大小,琼脂糖凝胶电泳法按照2015年版中国药典三部附录ⅳb,胶浓度为2%,核酸燃料gelred浓度为1%,电泳缓冲液为tae缓冲液(tris-edta-乙酸缓冲液,ph8.3),上述步骤(2)中获得的各样品的pcr反应溶液的上样量分别为10μl,dna分子量标记(dnamarker)上样量为5μl,电泳结束后,在凝胶成像仪上观察结果。若所述pcr扩增产物中含有101bp的dna片段,则所述待测阿胶存在驴源性成分;反之,则所述待测阿胶不存在驴源性成分。
实施例5鉴定方法
本实施例提供了一种利用实施例1的特异性引物或实施例2的试剂盒鉴定驴源性成分的方法,包括如下步骤:
(1)待测阿胶用ni-nta预处理
取10mg阿胶样品3,加10mllysisbuffer(nah2po425mm,nacl500mm,naoh调节ph至6.0),温浴烊化至样品融化完全,置于ni-nta预装柱内(填料量按照每1mlni-nta填料结合12mg蛋白计),于冰上震摇结合0.5h,收集穿透液,100℃蒸干,即得预处理样品。
(2)dna提取
取预处理样品采用takaraminibestdna试剂盒提取总dna,按照说明书操作,步骤同实施例5。
(3)pcr扩增
以步骤(2)中提取的dna为模板,采用如下引物进行pcr扩增:
上游引物:5′-aggagcaacggtcattaca-3′;
下游引物:5′-gggtggctttgtctactga-3′。
所述pcr反应体系包括:
dna模板,50ng/μl,1μl;
上游引物,10μm,1.5μl;
下游引物,10μm,0.5μl;
taqdna聚合酶,5u/μl,1.2μl;
dntps,10mm,0.25μl;
10×pcr扩增缓冲液,4.5μl;
无菌超纯水补至25μl。
扩增反应在pcr仪上进行,所述pcr扩增程序如下:
95℃预变性5min,95℃变性30s,48℃退火30s,72℃延伸30s,进行40个循环,然后继续72℃延伸5min,降温至4℃。
(4)取步骤(3)中的pcr扩增产物进行琼脂糖凝胶电泳,测得各样品的pcr扩增产物的大小,琼脂糖凝胶电泳法按照2015年版中国药典三部附录ⅳb,胶浓度为2%,核酸燃料gelred浓度为1%,电泳缓冲液为tae缓冲液(tris-edta-乙酸缓冲液,ph8.3),上述步骤(2)中获得的各样品的pcr反应溶液的上样量分别为10μl,dna分子量标记(dnamarker)上样量为5μl,电泳结束后,在凝胶成像仪上观察结果。若所述pcr扩增产物中含有101bp的dna片段,则所述待测阿胶存在驴源性成分;反之,则所述待测阿胶不存在驴源性成分。
对比例1鉴定方法
取阿胶样品1,按照实施例3的方法进行鉴定,与实施例3的区别仅在于阿胶样品1不经ni-nta预处理而是直接进行dna提取。
对比例2鉴定方法
取阿胶样品2,按照实施例4的方法进行鉴定,与实施例4的区别仅在于阿胶样品2不经预处理而是直接进行dna提取。
对比例3鉴定方法
取阿胶样品3,按照实施例5的方法进行鉴定,与实施例5的区别仅在于阿胶样品3不经预处理而是直接进行dna提取。
实验例1
利用紫外分光光度计法检测实施例4-5和对比例1-4步骤(2)提取的总dna的od260值,并计算od260/od280比值,根据od260/od280比值来考察所提取dna样品纯度,当od260/od280比值介于1.8~2.0之间时,说明样品纯度较高,当样品中如果含有蛋白质干扰,od260/od280比值会明显下降,结果如下表所示。
从上表可知,相比于对比例1-3来说,本发明实施例3-5提取的dna的od260/od2800比值明显提高,且均在介于1.8~2.0之间,说明本发明实施例3-5提取的dna的纯度明显提高,说明通过对阿胶样品进行预处理,提取的dna的浓度和纯度得到显著提高,且经pcr扩增可用于鉴定阿胶中是否存在驴源性成分。
此外,从图6中可以看出,m为dnamarker(50bp),条带1为实施例3的,条带2为实施例4,条带3为对比例5,条带4为对比例6,条带5为空白对照,从图中可以看出实施例3和4的阿胶样品具有特征谱带,明亮清晰,则证明所述阿胶样品中存在驴源性成分,而对比例5和6谱带模糊或不可见。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
sequencelisting
<110>北京同仁堂科技发展股份有限公司
<120>一种鉴定驴源性成分的特异性引物、试剂盒及鉴别方法
<130>ha201804183
<160>9
<170>patentinversion3.3
<210>1
<211>19
<212>dna
<213>人工合成
<400>1
aggagcaacggtcattaca19
<210>2
<211>19
<212>dna
<213>人工合成
<400>2
gggtggctttgtctactga19
<210>3
<211>19
<212>dna
<213>人工合成
<400>3
cccttagtcaatgcgtgtt19
<210>4
<211>19
<212>dna
<213>人工合成
<400>4
gcttgcgagtggtatgaag19
<210>5
<211>19
<212>dna
<213>人工合成
<400>5
atcatcacagccctggtaa19
<210>6
<211>20
<212>dna
<213>人工合成
<400>6
gggtgtagttgtctgggtct20
<210>7
<211>101
<212>dna
<213>人工合成
<400>7
aggagcaacggtcattacaaacctcctatcagcaatcccctacatcggtactacactcgt60
cgaatggatctgaggtggattctcagtagacaaagccaccc101
<210>8
<211>155
<212>dna
<213>人工合成
<400>8
cccttagtcaatgcgtgttctgactcttagtagcagacttactaacattaacatgaatcg60
gtggccaaccagtagaacacccatacgtaatcatcggccaactggcctcaatcctctact120
tctccctaattctcatcttcataccactcgcaagc155
<210>9
<211>211
<212>dna
<213>人工合成
<400>9
atcatcacagccctggtaatcgtccatctactattcctccacgaaacaggatccaacaac60
ccctcaggaatcccatctgacatagacaaaatcccattccacccgtactacacaattaaa120
gacatcctaggacttcttctcctagtcctactcctactaaccctagtattattctcccct180
gacctcctaggagacccagacaactacaccc211
- 还没有人留言评论。精彩留言会获得点赞!
- 一种特异性引物检测鼠源性成分的方法与制造工艺
- 一组用于驴源性的实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一组用于鸡源性实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 实时荧光PCR特异性检测肉苁蓉的引物及检测方法与制造工艺
- 小酸浆的分子特异性标记引物和检测方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 毛酸浆的分子特异性标记引物及鉴别方法与制造工艺
- 一种检测CYP2C9基因多态性的ARMS引物的制造方法与工艺
- 基于环介导等温扩增消光技术的玉米检测方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 毛酸浆的分子特异性标记引物及鉴别方法与制造工艺
- 一种检测CYP2C9基因多态性的ARMS引物的制造方法与工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 使用重叠非等位基因特异性引物和等位基因特异性阻断剂寡核苷酸的组合物进行等位基因特异性扩增的制造方法与工艺
- 一种利用线粒体D-loop区特异区域鉴定鼋的标记引物及方法
- 一种用于检测大豆拟茎点种腐病菌的特异性lamp引物组合物及其应用
- 一种针对膨胀污泥中Eikelboom Type 0092型丝状菌群特异性引物和方法
- 脯氨酸特异性蛋白酶基因及其应用