一种检测KRAS基因外显子2密码子12/13突变的试剂盒及其制备方法与流程
本发明涉及生物医学核酸检测试剂盒技术领域,具体为检测kras基因外显子2密码子12/13突变的方法及试剂盒的研发,同时提供其制备方法。
背景技术:
随着个体化医疗观念的不断深入,检测特定基因的特定位点突变已经成为指导靶向治疗用药的先决条件。酪氨酸激酶抑制剂(tki)是近年来肺部肿瘤治疗的热点靶向药物,与传统放化疗药物相比,可明显延长肿瘤患者生存期,改善生存质量。
肿瘤发生和发展受癌基因驱动。在众多癌基因中,ras基因家族在人类肿瘤中突变概率最高,被视为癌基因领域中的'珠穆朗玛峰'。ras突变肿瘤占人类所有恶性肿瘤的三分之一,其中kras作为ras基因家族中的主要亚型,促发人类多种致死性肿瘤,如肺癌、结肠癌和胰腺癌。然而,由于kras信号通路调控的复杂性以及kras突变肿瘤对临床药物的抵抗性,使得目前临床上仍无治疗kras突变肿瘤的有效药物和方法。kras突变型肿瘤是最具潜在靶向性的非小细胞肺癌分子亚型(nsclc)。在nsclc的病例中,kras突变主要发生在12和13号密码子。最常见的密码子变异约占kras突变型nsclc的39%,是kras-g12c突变。其他常见突变包括kras-g12v(18-21%)和kras-g12d(17-18%)变体。
目前基因变异的检测技术主要有直接测序法(亦称sanger测序法)和arms法。现分别简介于下:
测序法:测序法需要对测序样品扩增、纯化、序列分析,过程比较繁琐、耗时长,且对取材和技术要求比较高。聚合酶链式反应-单链构象多态性分析(pcr-sscp)pcr-sscp是一种比较经典的基因突变检测的方法,该方法可以对未知的突变进行检测。与测序法相比,pcr-sscp灵敏性更高,操作简单,无需特殊仪器,还可以对未知的突变进行检测。arms法也称扩增阻碍突变系统(amplificationrefractorymutationsystem,arms)、等位基因特性pcr(allelespecificpcr,aspcr)等,即等位基因特异性扩增法(allelespecificamplification,asa,选择性地扩增野生型或突变型基因。
上述现有技术存在以下问题:价格昂贵,操作复杂,且一般需要通过有创性组织活检,同时不易重复获取。研究表明,在血液、胸腔积液、唾液、粪便等样本中,会有少量混杂于大量野生型基因组dna中的突变肿瘤细胞dna。从这些低含量样本中检测突变的基因,需要找一种灵敏性高、特异性强、简便易行、结果判定简单的突变检测方法,用于癌症患者,特别是肺癌患者的靶向用药指导、高灵敏度早期复发监测,以及用药期间耐药性突变监测等。
技术实现要素:
本发明的目的在于提供一种检测kras基因外显子2密码子12/13突变的试剂盒,该试剂盒价格低廉,操作简单,可重复获取,且能同时检测多种突变类型,同时提供该试剂盒的检测方法,为个体化用药提供参考。
本发明以美国伯乐微滴式数字pcr为平台,选择kras基因2外显子区域进行探针和引物的设计。所选择的2外显子密码子12/13突变的位点在kras外显子2上c.5562-c.5566,此潜在的2外显子密码子12/13突变区域涵盖以下突变的类型见表1:
表1本发明可以检测的kras基因2外显子密码子12/13突变的类型
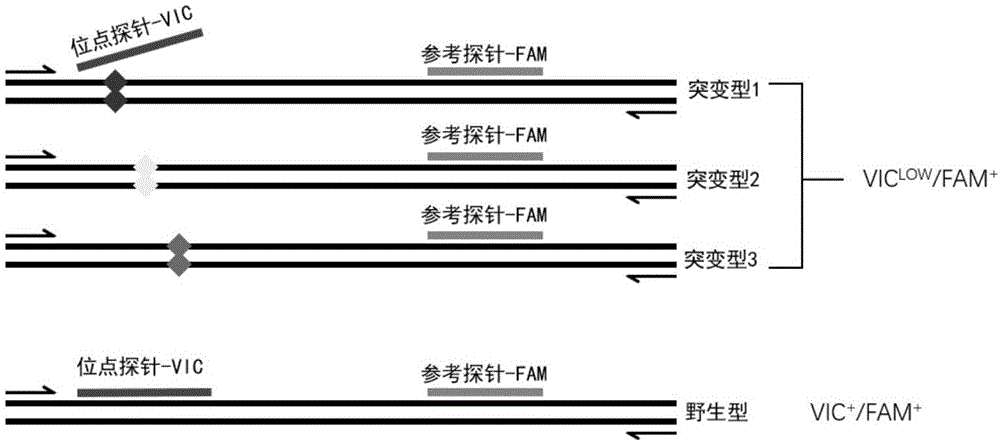
本发明的技术原理如图2所示,首先设计与kras基因2外显子密码子12/13突变的野生型序列为模板的taq-man探针,此探针标记以vic荧光基团,定义为位点探针。
本发明的技术原理如图1所示,在kras基因2外显子3’端非突变区域设计,以野生型序列为模板的taq-man探针,此探针标记以fam荧光基团,定义为参考探针。
本发明的技术原理如图1所示,若所测样本为野生型,则位点探针与参考探针均可与样本dna序列结合,探针上的vic与fam荧光基团均可被检测到相同或极其相近的信号强度;若样本出现上述任一kras基因2外显子密码子12/13的突变,vic标记的位点探针不能与样本dna结合,而fam标记的参考探针可与样本dna结合,因此可以检测到fam荧光信号,以及较弱的vic信号(kras基因2外显子密码子12/13杂合缺失突变),或检测不到vic信号(kras基因2外显子密码子纯合12/13缺失突变)。
本发明技术方案如下:
一种检测kras外显子2密码子12/13突变的试剂盒,包括:pcr扩增引物,taq-man探针,数字pcr预混液、微滴生成油、阴性对照和阳性对照;
所述的pcr扩增引物,包括反向pcr扩增引物和正向pcr扩增引物,可扩增含有kras外显子2上c.5522-c.5637(116-bpamplicon)序列如seqno.1所示;
所述的pcr扩增引物序列如下:
上游引物序列如seqno.2所示;
下游引物序列如seqno.3所示;
参考探针序列如seqno.4所示;
位点探针序列如seqno.5所示。
所述的位点探针及参考探针的5’端连接有荧光基团,3’端连接有猝灭基团。
所述的荧光基团选自6-羧基荧光素、六氯-6-羧基荧光素、fam、cy5、cy3或vic;
所述的猝灭基团选自6-羧基四甲基若丹明、bhq1、bhq2或mgb。
所述的数字pcr预混液主要成分包含taqdna聚合酶、dntp、mgcl2、pcrbuffer、pcr反应增强剂、稳定剂。
所述的上游引物和下游引物的使用浓度为5μm~15μm,在最终反应体系中的浓度为400nm~1200nm。
所述的位点探针和参考探针的使用浓度为5μm~25μm,在最终反应体系中的浓度为100nm~500nm。
所述的阴性对照来源于携带kras基因野生型的非小细胞肺癌细胞h1975;阳性对照来源于携带kras基因2外显子密码子12/13突变的阳性细胞人结肠癌细胞sw480或hct-116。
上述的试剂盒的制备方法,包括以下步骤:
(1).dna提取:提取含有kras基因2外显子密码子12/13突变的阳性细胞人结肠癌细胞sw480或hct-116和kras基因野生型细胞h1975;
(2).数字pcr反应液制备;
(3).pcr反应微滴制备:将pcr反应液转移至微滴发生卡,在油孔中加入微滴生成油,制备微滴;
(4).pcr扩增:退火后,然后扩增反应;
(5).检测及计算突变率。
所述步骤(2)包括:将待检测样品、pcr扩增上下游引物、无酶水、kras基因2外显子密码子12/13突变的位点探针和参考探针以及数字pcr预混液混合混合制备得到数字pcr混合液。
所述的步骤(4)的扩增反应过程为:93~97℃预变性3~15分钟;93~97℃变性5~50秒,55℃~63℃退火/延伸60~120秒,,共进行20~60个循环,95℃~100℃酶失活8~16分钟,16℃无限循环。
本发明具有积极的效果
(1).成本低
目前肽核酸(peptidenucleicacid,pna)也可用于多位点突变的检测。它与dna模板结合能力高于普通寡核苷酸,若pna与模板正确配对时可阻止聚合酶链反应,野生型模板无法扩增;若发生错配,则结合能力迅速下降,此时可用来扩增突变模板。这种检测方法有着难以避免的劣势和局限性。一方面,在此方法中需要加入pna的探针用以与野生型dna结合,这就增加了繁琐的操作步骤;且pna与dna结合的效率并非100%,这极易造成检测结果偏差,难以避免出现假阴性或假阳性;另一方面,pna造价比较高,成倍的增加检测成本,难以大范围临床推广应用。
(2).一次检测kras最常见的密码子12和13位点的多种缺失信息
本发明基于微滴式数字pcr技术平台,由于其仅须一次pcr反应即可同时检测kras最常见的密码子12和13位点的多种缺失类型,可明显减少试剂、耗材的消耗,单次检测的成本仅为其他微滴式数字pcr的1/8。本发明的kras外显子2密码子12/13突变检测技术与一般定量相比,不用设置标准品,也无需设置质控品。一般检测技术是定性检测,突变检出率较低,本发明的技术可以达到真正意义的绝对定量,克服了qpcr等技术无法精确定量的缺点。
(3).操作简便
本发明基于微滴式数字pcr技术发展起来,它结果判读方式是数据信息,可以借助软件完全自动化的进行结果判读,从而加快了数据分析的速度,也减少了产生假阴性和假阳性判读的可能。它结果描述的方式是绝对定量的方式,可以给出样本所携带某种特异基因变异dna的绝对数量和比例。而且准确率高,即使在突变样本含量较低的情况下,定量分析的误差也很小。
(4).灵敏度极高
以前技术方法的灵敏度一般在1%-50%之间,而我们提出的技术方法对基因变异的检测灵敏度提升非常明显,为1/1000。可以在非常高背景的野生dna中检测出极其微量的携带基因变异的dna。
附图说明
图1基于数字pcr法检测kras外显子2密码子12/13突变原理示意图;
图2不同温度下,kras基因2外显子密码子12/13突变数字pcr的扩增效率及特异性检测;
图3在63℃kras基因2外显子密码子12/13突变数字pcr的扩增效率及特异性检测;
图4特异性检测:加入不同拷贝数的外源性dna,检测特异性;
图5特异性检测:加入46,600拷贝数的外源性dna,检测特异性;
图6灵敏度检测:4.81%突变率dna荧光检测结果;
图7灵敏度检测:3%突变率dna荧光检测结果;
图8灵敏度检测:1.82%突变率dna荧光检测结果;
图9灵敏度检测:0.64%突变率dna荧光检测结果;
图10灵敏度检测:0.16%突变率dna荧光检测结果;
图11灵敏度检测:0.08%突变率dna荧光检测结果(未检测到);
图12实例分析:kras基因2外显子g13a突变的石蜡包埋组织切片标本检测结果。
具体实施方式
下面通过实施例对本发明进行具体的描述,同样应理解,以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容作出的一些非本质的改进和调整均属于本发明的保护范围。下述示例具体的工艺参数等也仅是合适范围中的一个示例,即本领域技术人员可以通过本文的说明做合适的范围内选择,而并非要限定于下文示例的具体数值。
下述实施例中,若非特意表明,所用的试剂均为分析纯,所用试剂均可从商业渠道获得。文中未注明具体条件的实验方法,通常按照常规条件如j·萨姆布鲁克等编著的科学出版社2002年出版的《分子克隆实验指南》一书中所述的条件,或按照制造商所建议的条件。除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
本发明的序列均来自ncbi,根据ncbi公布的kras基因2外显子野生型及密码子12/13突变型序列设计及合成高特异性、高灵敏性的探针、引物。dna来源主要有人结肠癌细胞系sw480或hct-116的基因组dna,此细胞分别为kras基因2外显子g12v和g13d突变,可作为阳性对照;人非小细胞肺癌细胞系h1975的基因组dna,此细胞为kras基因野生型,可作为阴性对照;kras基因2外显子g13a突变的石蜡包埋组织切片标本用于实例验证分析。
一)试剂盒构成:
表2试剂盒成分表
本实施例的egfr基因突变检测试剂盒,包括数字pcr预混液、微滴生成油、taq-man探针、pcr引物、阴性对照和阳性对照。上述表2试剂盒成分说明如下:
数字pcr预混液主要成分包含taqdna聚合酶、dntp、mgcl2、pcrbuffer、pcr反应增强剂、稳定剂等(由bio-rad公司提供,货号:1863023)
微滴生成油主要成分是矿物油,(由bio-rad公司提供,货号:1863005),主要作用是微滴化处理过程中将pcr产物形成油包水微液滴。
taq-man探针:用于识别egfr外显子19缺失突变的taqman探针,分别为上述位点探针和参考探针,是连接荧光基团和猝灭基团的核苷酸,序列见表3
pcr引物:为反向pcr扩增引物和正向pcr扩增引物,可扩增含有kras外显子2上c.5522-c.5637(116-bpamplicon)
ctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatccaacaataga序列的片段。序列见表3
表3:引物探针序列
kras基因2外显子密码子12/13突变上游引物和下游引物的使用浓度为5μm~15μm,在最终反应体系中的浓度为400nm~1200nm,优选的kras基因2外显子密码子12/13突变上游引物和下游引物的使用浓度为10μm,在最终反应体系中的浓度为800nm,
kras基因2外显子密码子12/13突变的位点探针和参考探针的使用浓度为5μm~25μm,在最终反应体系中的浓度为100nm~500nm,优选的kras基因2外显子密码子12/13突变的位点探针和参考探针的使用浓度为10μm,在最终反应体系中的浓度为200nm,
阴性对照来源于携带kras基因野生型的非小细胞肺癌细胞h1975。
阳性对照来源于携带kras基因2外显子密码子12/13突变的阳性细胞人结肠癌细胞sw480或hct-116。
二)试剂盒的使用方法:
本实施例的kras基因2外显子密码子12/13突变的具体检测步如下:
1.dna提取
细胞基因组dna的提取:培养含有kras基因2外显子密码子12/13突变的阳性细胞人结肠癌细胞sw480或hct-116和kras基因野生型细胞h1975。用tiangen公司提供的血液/细胞/组织基因组dna提取试剂盒(货号:dp304),按照实际说明书提取基因组dna.然后通过ksp-3700测定其dna浓度,确定加样量。
石蜡包埋组织切片标本游离dna的提取:采用存在kras基因2外显子g13a突变的石蜡包埋组织切片标本,用qiagen公司提供的石蜡包埋组织游离核酸提取试剂盒(货号:56404),按照试剂说明书提取游离dna。然后通过thermofisher公司qubit3.0核酸蛋白荧光定量仪,测定其游离dna的浓度和纯度,控制加样量。
数字pcr反应液制备
将待检测样品、pcr扩增上下游引物、kras基因2外显子密码子12/13突变的位点探针和参考探针以及数字pcr预混液混合,不足22ul反应体系的用无酶水补齐,制备得到数字pcr混合液。
表4:检测kras基因2外显子密码子12/13突变的ddpcr
3.pcr反应微滴制备
将配制好并混合均匀的22μlddpcr反应液,转移至微滴发生卡(dg8cartridge)中间一排样本孔中,避免孔底部产生气泡;随后在油孔中加入70μl微滴生成油(dropletgenerationoil),安装胶垫;将装载好的微滴发生卡放入qx200tm微滴生成器制备微滴。每个微滴发生卡一次可同时完成8个样品的微滴制备,一般在2分钟内完成。本实验反应体系通过。微滴成器形成上百万油包水微滴在进行pcr反应,每个微滴至少含有一个或者不含待检核酸靶分子。
退火温度设定
温度梯度设定依次为64℃,63.7℃,63℃,61.8℃,60.4℃,59.2℃,58.4℃,58℃,退火时间2min。(见图2)。各孔加入同等量的h1975dna(kras野生型)。观察不同温度下,kras基因2外显子密码子12/13突变数字pcr的扩增效率及特异性比较。63℃时数字pcr的扩增效率及特异性最好。故确定最佳tm63℃(见图3)。
扩增
将制得的每个样品的微滴分别转移至96孔pcr板中对应的反应孔中。用铝膜热封(180℃,5sec),于普通pcr仪上进行扩增。pcr反应程序为:93~97℃预变性3~15分钟;93~97℃变性5~50秒,58℃~64℃退火/延伸60~120秒,,共进行20~60个循环,95℃~100℃酶失活8~16分钟,16℃无限循环。
优选地,数字pcr扩增条件为:95℃预变性10分钟;94℃变性30秒,63℃退火/延伸120秒,共进行40个循环,98℃酶失活10分钟,16℃无限循环。
表5:pcr扩增程序设定
6.微滴检测
将pcr扩增后的96孔板放入qx200tm微滴式数字pcr仪的微滴读取仪中,并在软件quantasoft上正确选择试剂种类,并设定实验类型为red,同时检测fam和vic的荧光信号。仪器自动分析每个样品的每个微滴的荧光信号,含有荧光信号的标记为1,不含荧光信号的标记为0。然后,通过泊松分布公式自动分析,quantasoft软件会根据阳性微滴的比例获得pcr反应体系中的起始模板dna拷贝数浓度,并给出突变百分比。
结果判定
对pcr扩增反应后的产物进行信号收集,根据荧光信号确定待测样品kras基因2外显子密码子12/13位点是否突变,含未发生突变的野生型dna模板的微反应微滴中,同时出现fam和vic荧光信号,记为一个信号数;含突变的dna模板的微反应微滴中,仅出现fam荧光信号。
突变率计算
2外显子的密码子12/13位点突变率的计算公式如下:
(三)试剂盒的灵敏度和特异性:
特异性检测:将sw480dna(kras基因2外显子g12v突变型)按照不同拷贝数稀释,拷贝数依次为600拷贝,1250拷贝,2500拷贝,5000拷贝,10000拷贝,16500拷贝,25000拷贝,46600拷贝(见图4),当拷贝数为46600拷贝时,仍能明显检测其突变(见图5),证实此发明具有极好的特异性。
灵敏度检测:将sw480dna(kras基因2外显子g12v突变型)和h1975dna(kras基因野生型)按照不同拷贝数进行稀释互掺。当突变率为0.16%时,仍能检测其阳性突变,继续稀释,未能检测其阳性突变(见图6~11),证实此发明具有极好的敏感性。
四)应用例:
1.石蜡包埋组织中ctdna检测
①kras基因2外显子g13a突变的石蜡包埋组织切片标本(emqn购进),置入1.5mlep管中,加入1ml二甲苯溶解石蜡组织。
②用qiagen公司提供的石蜡包埋组织游离核酸提取试剂盒(货号:56404),按照试剂说明书提取游离dna,用30ul水溶。
③ctdna经qubit3.0测定其浓度和纯度后,确定加样量8ul。
结果如图12所示,石蜡包埋组织切片标本游离dna拷贝数为:260拷贝;kras基因2外显子g13a突变比率为:44.77%。
序列表
<110>宋现让
<120>一种检测kras基因外显子2密码子12/13突变的试剂盒及其制备方法
<160>5
<170>siposequencelisting1.0
<210>1
<211>116
<212>dna
<213>人工序列(artiartificialsequence)
<400>1
ctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgcc60
ttgacgatacagctaattcagaatcattttgtggacgaatatgatccaacaataga116
<210>2
<211>26
<212>dna
<213>人工序列(artiartificialsequence)
<400>2
tctattgttggatcatattcgtccac26
<210>3
<211>29
<212>dna
<213>人工序列(artiartificialsequence)
<400>3
ctgaaaatgactgaatataaacttgtggt29
<210>4
<211>20
<212>dna
<213>人工序列(artiartificialsequence)
<400>4
agctgtatcgtcaaggcact20
<210>5
<211>15
<212>dna
<213>人工序列(artiartificialsequence)
<400>5
cctacgccaccagct15
- 还没有人留言评论。精彩留言会获得点赞!