一类靶向治疗耐甲氧西林金葡菌、金葡菌及超级细菌感染的抗菌药物ASC的合成及应用的制作方法
本发明涉及一类靶向治疗耐甲氧西林金葡菌(mrsa)及金葡菌(sau)感染且协同抗生素治疗超级细菌感染的抗菌药物asc的合成方法及应用,属药物化学领域。
背景技术:
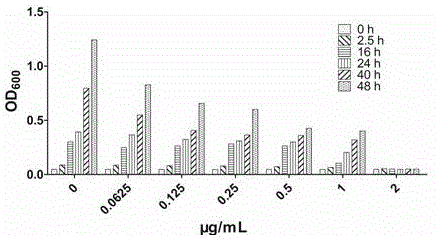
:金属β-内酰胺酶(metallo-β-lactamases,mβls)介导的“超级细菌”耐药导致几乎所有临床上使用的抗生素失效。因而,对产mβls耐药细菌的监测与抑制是世界卫生组织(who)、各国政府及医药行业目前密切关注的热点。抗生素在临床、农业和畜牧业方面的过度使用,导致大量的耐药细菌产生,使得在选择对付这些耐药细菌的药物时存在巨大的选择压力。近年来,临床上分离到的产mβls的菌种如铜绿假单胞菌、金黄色葡萄球菌、鲍曼不动杆菌以及炭疽芽胞杆菌等明显增加,且其耐药性越来越强,以至发展到携带ndm-1的“超级细菌”自2010年起在全球蔓延,迄今尚无任何药物对付。回顾抗生素的耐药历史,一个新抗生素的投入使用,长则4年,短则不到1年就出现了相应的耐药细菌。可见,新抗生素的开发速度远远跟不上细菌耐药发生的步伐。故而,依靠不断研发新抗生素来对付耐药细菌的路走不通。应对抗生素耐药的理想策略是,研究和发展mβls的抑制剂,以其与抗生素协同来抑制。自青霉素被发现以来,β-内酰胺类抗生素已经发展成为临床上的主力抗菌药物,目前临床上使用的抗菌药物约60%为β-内酰胺类抗生素。β-内酰胺类抗生素是广谱抗菌药物,也是治疗细菌感染最成功的药物之一。β-内酰胺类抗生素有上百种,按结构其主要分为四类:青霉素类、碳青霉烯类、头孢菌素类和单酰胺环类。β-内酰胺类抗生素分子均含有一个β-内酰胺环,其侧链的结构变化形成了不同的抗生素。β-内酰胺类抗生素的作用机制通常是抑制转肽酶的合成,从而阻止细胞壁的合成。抗生素在临床、农业和畜牧业方面的过度使用,导致大量的耐药细菌产生。细菌对β-内酰胺类抗生素产生耐药性的机理有多种,其中最重要的机理是产生β-内酰胺酶。细菌通过生产β-内酰胺酶催化水解β-内酰胺类抗生素的β-内酰胺环而呈现出耐药性。目前,已经确定的β-内酰胺酶有1000多种,这些酶被归类为a,b,c和d四组。其中a、c和d组酶被称之为丝氨酸β-内酰胺酶(serine-β-lactamases,sβls),其丝氨酸残基扮作亲核基团进攻β-内酰胺环之羰基碳以水解抗生素而发挥作用。而b组酶,即金属β-内酰胺酶(metallo-β-lactamases,mβls),其活性的正常发挥通常依赖于zn(ii)离子。根据基因序列和与zn(ii)离子结合方式的不同,mβls被进一步归类为b1、b2、b3三个亚组。mβls水解几乎所有已知的抗生素,但是,迄今临床上还没有任何药物可以用来处理携带mβls的超级耐药细菌。鉴于β-内酰胺酶介导的细菌耐药对人类健康的威胁日益严重,人们做了大量的研究在寻找β-内酰胺酶的抑制剂。1972年,丝氨酸β-内酰胺酶的抑制剂克拉维酸被发现。1984年,克拉维酸作为第一个丝氨酸β-内酰胺酶的抑制剂被fda批准用于临床。克拉维酸分子含有一个β-内酰胺环,可与靶蛋白活性中心结合形成一个酰基酶复合物,然后重排形成酰胺中间体而不可逆性抑制酶的活性。随后,克拉维酸与阿莫西林联用开发出阿莫西林-克拉维酸钾片(也称为奥格门丁),成功地实现了β-内酰胺酶抑制剂与β-内酰胺抗生素的协同抑菌效能。这是人类历史上第一次使用协同理念治疗耐药细菌感染。但遗憾的是,克拉维酸与抗生素的联用却对产金属β-内酰胺酶的耐药细菌无效。金黄色葡萄球菌(staphylococcusaureus)为革兰氏阳性菌,属于厚壁菌,常见于人体的鼻腔、呼吸道和皮肤组织。近年来,金葡菌已成为全球主要致病菌之一,部分原因是因为它在公共场所的广泛传播,增加了反复传染的风险。另外,金葡菌对抗生素的耐药性越来越明显。金葡菌可产生多种酶,例如保护细菌细胞的凝固酶、分解透明质酸的透明质酸酶、降解脂肪的脂肪酶、溶解纤维蛋白的葡萄菌激酶、破坏dna的脱氧核桃核苷酸酶,以及造成金葡菌对抗生素耐药的β-内酰胺酶等。金葡菌可以释放出青霉素酶,该酶可以水解抗生素的β-内酰胺环,使得此类抗生素失去对细菌的破坏作用。对甲氧西林、苯唑西林、头孢西丁呈耐药性或meca基因呈阳性的金黄色葡萄球菌被定义为耐甲氧西林金黄色葡萄球菌(methicillin-resistantstaphylococcusaureus,mrsa)。自首株mrsa检出以来的40余年,mrsa感染在世界各地一直呈上升趋势。美国医院内感染监测系统(nnis)报道,1975年182所医院mrsa占金葡菌感染总数的2.4%,1997年上升至24.8%。90年代后,国内京、沪等地大型医院mrsa分离率均超过金葡菌感染总数的50%以上。抗生素的滥用导致了mrsa多重耐药现象日益严重,例如对β-内酰胺类、大环内酯类、四环素类、氨基糖苷类、氟喹诺酮类和磺胺类等多种抗生素均产生了不同程度的耐药。糖肽类抗生素万古霉素是治疗mrsa感染的最后一道防线,但现已出现耐万古霉素金黄色葡萄球菌(vancomycinintermediary-resistantstaphylococcusaureus,visa)和耐万古霉素金黄色葡萄球菌(vancomycinresistantstaphylococcusaureus,vrsa)。近年来,对利奈唑胺和达托霉素等抗mrsa药物也相继发现其耐药菌。因此,寻找有效抵抗mrsa的新药已经迫在眉睫。mrsa的耐药机制主要包括:(1)大量表达由meca基因编码产生青霉素结合蛋白(pbp2a)。β-内酰胺类抗生素可抑制转肽酶pbp2的活性,但在抗菌药物存在时,pbp2a易与pbp2形成复合物而补充或代偿抗菌药物抑制pbp2的功能,从而继续维持细菌细胞壁的合成;(2)产生β-内酰胺酶,催化水解β-内酰胺类抗生素的β-内酰胺环而呈现出耐药性。综上,β-内酰胺酶介导的细菌耐药对人类健康的威胁日益严重。尽管克拉维等丝氨酸β-内酰胺酶抑制剂联用抗生素对革兰氏阴性耐药菌具有良好的抑制效果。然而,其对携带金属β-内酰胺酶的耐药细菌却无效。迄今,临床上没有任何药物可以用来处理携带金属β-内酰胺酶的超级耐药细菌。技术实现要素:本发明的目的是提供一类兼具抗菌试剂和细菌耐药靶酶抑制剂的靶向治疗金黄色葡萄球菌感染的新型抗菌药物asc及其合成方法。为了实现上述目的,本发明通过下述技术方案来实现:结构通式(i)所示化合物:r1为r为-nh2、-cho、-cooh、-oh、-cl或-br;r2为-nh2或-h;r3为x为n或s。进一步地,所述的如式(ⅰ)所示的化合物为如下所示的任一化合物:上述化合物的制备方法,包括以下步骤:(1)在三乙胺存在下,a1在乙腈中用r3-cl酰化制备得a2;(2)a2在丙酮中加入碘化钠碘代反应制得a3;(3)在n-甲基吗啉存在下,a3在氯仿中与如下巯基唑类化合物反应制得a4;(4)在苯甲醚存在下,a4在二氯甲烷中用三氟乙酸脱保护基得到asc;具体描述,上述步骤(1)中,将a1与2,6-二甲基吡啶溶解于乙腈冷却至0℃,滴加酰氯,然后加入等摩尔比的三乙胺,搅拌过夜后升至室温;减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,用饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏得a2。上述步骤(2)中,将a2与碘化钠溶于丙酮,室温搅拌,除去溶剂后所得残留加入水、乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏制得a3。上述步骤(3)中,在n-甲基吗啉存在下,tlc跟踪,待a3与b4在氯仿中反应结束后升温至室温,除去溶剂,柱硅胶分离纯化得到a4。上述步骤(4)中,在苯甲醚存在下,a4在二氯甲烷中用三氟乙酸脱保护基,待反应结束后,减压蒸馏除去溶剂,所得残留用乙醚洗涤得asc。上述化合物在制备协同抗生素靶向性治疗金黄色葡萄球菌感染药物中的应用。以上化合物作为药物,是一种细菌耐药靶蛋白金属β-内酰胺酶的抑制剂,可协同β-内酰胺类抗生素治疗金属β-内酰胺酶介导的超级耐药菌,并治疗mrsa感染。所述的β-内酰胺类抗生素为青霉素类、头孢菌素类及碳青霉烯类抗生素中的一种或多种。本发明至少提供了该化合物协同头孢唑林、亚胺培南等药物的测试。本发明的有益效果为:本发明提供的asc不仅本身是一类抗菌试剂,而且还是一类β-内酰胺类抗生素耐药靶蛋白金属β-内酰胺酶的广谱型抑制剂。本发明所述β-内酰胺类抗生素,是指分子中含有由四个原子组成的β-内酰胺环类抗生素,包括常见的青霉素类、头孢菌素类及碳青霉烯类抗生素中的一种或多种。asc可协同β-内酰胺类、氨基糖苷类和四环素类三类7-8种抗生素(青霉素g、氨苄西林、头孢唑林钠、头孢曲松钠、头孢噻肟钠、亚胺培南、卡那霉素、四环素等)靶向性治疗金黄色葡萄球菌的感染。联用1μg/ml剂量的asc,使得这些抗生素的活力提高4-128倍。asc与氨苄西林展现出最佳协同抑菌效能,即联用1μg/ml剂量的asc,使用目前临床上剂量1/128的氨苄西林就可灭活金黄色葡萄球菌;一个2μg/ml剂量的asc-na几乎100%地治疗金黄色葡萄球菌的感染,其效能维持至少48小时以上。asc可协同头孢唑林靶向抑制携带质粒pet-26-l1、pet-26-ndm-1、pet-26-vim-2、pet-26-ccra或pet-26-imis的超级耐药细菌e.coli。联用剂量64μg/ml的anb1h、anb0h、anb04n或ana2c,使得先锋ⅴ治疗产ndm-1的超级耐药细菌e.coli-dh10b的活力恢复了32倍。联用8μg/ml的asc-na、anb04o或ana2c,使得先锋ⅴ治疗产ccra的耐药菌e.coli-bl21的活力提高了32倍。联用16μg/ml的asc-nb或aba2c,使得先锋ⅴ治疗e.coli的活力恢复了64倍。此外,asc可有效治疗mrsa的感染,其中anb04n的治疗效果最佳(mic=2μg/ml),要比苯唑西林灭活mrsa的效力强达4倍。asc-nb对mrsa展现剂量依赖性灭活性能,一个剂量为16μg/ml的asc-nb几乎100%地灭活mrsa,其灭菌效能维持48小时以上。动物(小鼠)实验表明,asc-nb极有效地治疗小鼠感染的mrsa。一个给药量为35mg/kg的asc-nb,使得小鼠心脏和肺部感染的mrsa几乎全部被杀死,一周后仍无细菌繁殖;同剂量的asc-nb,也使得小鼠的肝、脾和肾组织中mrsa的定殖量显著降低。asc-nb治疗mrsa的效能随给药浓度和用药时间的不同而呈依赖性变化。显然,asc-nb对小鼠感染的mrsa有很好的治疗效果,这预示:asc具有成为临床治疗mrsa感染的潜在药物的可能性。本专利填补了一项世界空白。附图说明图1是asc-na对金黄色葡萄球菌灭活效能随时间及其浓度的变化;图2为先锋ⅴ灭活mrsa的剂量及时间依赖性变化;图3为asc-nb灭活mrsa的剂量及时间依赖性变化;图4为小鼠在空白、感染mrsa后及分别给药(asc-nb或苯唑西林)后器官组织中mrsa定殖量的变化。具体实施方式下面通过具体实例进一步说明本发明的实施方式,但并不用于限制本发明的实施范围。中间产物b4的合成路线:(1)将b1与水合肼按1:1的摩尔比混合后升温至80℃,反应回流8-12h后冷却至室温,在冷却过程中有大量白色固体b2形成。(2)将b2溶于足够量的乙醇中,室温搅拌至澄清,加入摩尔比为1:1的硫氰酸铵之后再加入浓盐酸,升温至80-100℃回流12-16h获得b3。(3)将b3溶于氢氧化物钠溶液中后升温至100-160℃,搅拌回流后用盐酸酸化至ph=5-6,所生成的白色固体用水洗至中性,干燥后得b4。实施例1本发明asc-nb结构式如下:上述asc-nb制备按以下步骤进行:1.中间产物c4的制备步骤如下:(1)将c1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体c2生成;1hnmr(400mhz,dmso-d6):δ12.52(s,1h),10.07(s,1h),7.79(d,1h),7.37(t,1h),6.87(dd,2h),4.65(s,2h)。(2)将c2溶于足够量的乙醇中,室温搅拌至澄清,加入等摩尔比的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应结束后冷却至室温,过滤除去不溶物,所得滤液减压蒸馏、干燥获得c3。(3)将c3溶于氢氧化物钠溶液升温至100-160℃回流,反应完毕后,用盐酸酸化至ph=5-6,所得大量白色固体用水洗至中性,真空干燥得到c4;1hnmr(400mhz,dmso-d6):δ13.70(s,1h),7.62(d,1h),7.79(d,1h),7.34(t,1h),6.99(dd,2h),6.92(t,1h)。2.中间产物c5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入c4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得c5;1hnmr(400mhz,cdcl3):δ7.32(d,j=8.5hz,9h),6.88(d,j=8.7hz,3h),6.11(d,j=9.1hz,1h),5.83-5.77(m,1h),5.17(s,2h),4.91(d,j=4.8hz,1h),4.29(s,2h),3.80(s,3h),3.64(d,j=8.2hz,2h),3.48(s,2h)。3.asc-nb的制备步骤如下:将c5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得asc-nb;1hnmr(400mhz,meod):δ7.32(d,j=4.7hz,7h),7.15(d,j=8.5hz,2h),6.87(d,j=8.5hz,2h),6.13(s,1h),5.45(d,j=3.8hz,1h),5.29(dd,j=8.6,3.7hz,1h),4.67(s,1h),3.82(s,1h),3.79(s,1h),3.59(s,1h),3.56(s,1h).13cnmr(101mhz,meod):δ154.92,154.57,154.16,151.85,145.58,134.59,133.98,130.32,126.38,125.51,123.65,122.83,122.57,120.16,115.80,71.41,71.38,71.34,70.30,68.51,46.71,9.77.hrms[m-h]-(m/z)forc24h20n5o5s2:calcd.522.0900,obsd.522.0930。实施例2本发明asc-na的结构式如下:1.中间产物d3的制备步骤,(1)按1:1摩尔比将d1与氨基硫脲在乙腈中混合并回流2h,冷却后所得固体用乙醇反复洗涤得中间产物d2。(2)将d2溶于氢氧化钠溶液,回流过夜后用稀盐酸调ph至酸性得到d3;1hnmr(400mhz,cdcl3):δ3.26(s,1h),2.53(t,j=5.9hz,2h),2.31(t,j=5.5hz,2h),1.89(p,j=5.7hz,2h)。2.中间产物d4的制备步骤,(1)将a1与2,6-二甲基吡啶溶解于乙腈,0℃下滴加1.5当量的酰氯2h,然后加入等当量的三乙胺,搅拌过夜后升至室温。tlc跟踪,待反应完成后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液和饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥,抽滤,减压馏除去溶剂,硅胶柱分离纯化得到a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥,减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃并在氮气保护下,缓慢加入d3,搅拌2h后升至室温继续反应。tlc跟踪,待反应完毕后除去溶剂,柱硅胶分离纯化制得d4;1hnmr(400mhz,cdcl3):δ8.02(s,1h),7.35-7.16(m,9h),6.90-6.77(m,2h),5.73(s,1h),5.15(d,j=5.9hz,2h),4.89-4.81(m,1h),4.27(d,j=13.7hz,1h),3.94(d,j=13.6hz,1h),3.74(s,3h),3.60(s,2h),3.45(d,j=18.4hz,1h),2.77(t,2h),2.34(t,2h),2.06-1.92(m,2h)。3.asc-na的制备步骤,将d4溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h。tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤获得asc-na;1hnmr(400mhz,meod):δ7.30(s,2h),7.29(d,j=1.4hz,2h),7.26-7.20(m,1h),5.67(d,j=4.8hz,1h),5.02(d,j=4.7hz,1h),4.38(d,j=13.6hz,1h),4.02(d,j=13.6hz,1h),3.84-3.74(m,2h),3.60–3.56(m,2h),3.54(d,j=7.0hz,1h),3.48(q,j=7.0hz,1h),2.83(dd,j=14.7,7.1hz,2h),2.37(t,j=7.3hz,2h),2.01(p,j=7.4hz,2h),1.17(m,2h).13cnmr(101mhz,meod):δ175.17,173.24,164.72,163.37,158.84,135.09,129.36,128.87,128.22,126.66,125.45,113.43,59.23,57.68,41.80,34.83,32.44,27.18,25.02,22.58.hrms[m-h]-(m/z)forc22h22n5o6s2:calcd.516.1016,obsd.516.1051。实施例3本发明asc-sb结构式如下:1.中间产物e4的制备步骤,(1)按1:1摩尔比将e1和水合肼混合,升温至80-100℃回流8-10h。待反应完成后冷却至室温,冷却后获得大量的白色固体e2。(2)将e2溶于足够量的乙醇,室温搅拌至澄清后加入等当量的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应完成后冷却至室温,过滤除去不溶物,滤液减压旋干,真空干燥获得e3。(3)将e3溶于少量浓盐酸,搅拌回流。待反应完成后将反应混合物倾入冰水中,有大量黄色固体产生。滤饼用水洗至中性再溶于氢氧化钠,滤去不溶物,滤液用稀盐酸调至ph=5-6,静置、抽滤、干燥得到e4;1hnmr(400mhz,cdcl3):δ7.30-7.21(m,5h),7.15(ddt,j=9.3,7.2,2.4hz,1h),3.81(s,2h),1.65(s,1h)。2.中间产物e5的制备步骤(1)将a1与2,6-二甲基吡啶溶于乙腈,0℃按1:1.5摩尔比滴加酰氯2h,然后加入等当量的三乙胺,搅拌过夜后升至室温。tlc跟踪,待反应完成后,减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,用饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥,抽滤,减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠室温搅拌2.5h。乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥,减压蒸馏制得a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃并在氮气保护下2h内缓慢加入e4,然后升至室温继续搅拌。tlc跟踪,待反应完毕后,除去溶剂,柱硅胶分离纯化得到e5;1hnmr(400mhz,cdcl3):δ7.26(d,j=2.3hz,3h),7.22-7.21(m,2h),7.20-7.15(m,7h),6.77(d,j=8.6hz,2h),6.71(d,j=9.0hz,1h),5.68(dd,j=9.0,4.9hz,1h),5.11(s,2h),4.77(d,j=4.9hz,1h),4.52(d,j=13.5hz,1h),4.24(s,2h),4.05–3.98(m,2h),3.71(s,1h),3.69(s,3h),3.53(s,2h)。3.asc-sb的制备步骤将e5溶于干燥的二氯甲烷,0℃加入苯甲醚和三氟乙酸并搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤获得asc-sb;1hnmr(400mhz,meod):δ7.38–7.37(m,2h),7.31(s,3h),7.30(s,5h),5.68(d,j=4.8hz,1h),5.51(d,j=4.3hz,1h),5.44(s,1h),5.19(d,j=5.9hz,1h),4.39(s,2h),3.77(s,2h),3.61(m,2h),3.59(s,2h).13cnmr(101mhz,meod)δ167.43,165.35,164.13,163.62,158.25,135.73,135.46,128.87,128.84,128.35,127.12,126.25,124.17,60.01,57.80,47.05,38.88,32.70,26.69.hrms[m-h]+(m/z)forc25h21n4o4s3:calcd.537.0719,obsd.537.0762。实施例4本发明anb1h结构式如下:上述anb1h制备按以下步骤进行:1.中间产物f4的制备步骤如下:(1)将f1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体f2生成;1hnmr(400mhz,dmso-d6):δ9.26(s,1h),7.28(m,5h),4.25(s,2h),3.37(s,2h)。(2)将f2溶于足够量的乙醇中,室温搅拌至澄清,加入等摩尔比的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应结束后冷却至室温,过滤除去不溶物,所得滤液减压蒸馏、干燥获得f3。(3)将f3溶于氢氧化钠溶液后升温至100-160℃回流,反应完毕后,用盐酸酸化至ph=5-6,所得大量白色固体用水洗至中性,真空干燥得到f4;1hnmr(400mhz,dmso-d6):δ7.36(dq,j=7.5,1.3hz,2h),7.29(t,j=7.5hz,2h),7.15(tt,j=7.3,2.0hz,1h),3.91(s,2h)。2.中间产物f5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入f4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得f5;1hnmr(400mhz,cdcl3):δ7.39-7.27(m,10h),7.22(d,j=7.4hz,2h),6.87-6.83(m,2h),6.27(d,j=9.0hz,1h),5.74(s,1h),5.21(d,j=11.8hz,1h),5.12(d,j=11.8hz,1h),4.80(d,j=4.8hz,1h),4.34(d,j=13.9hz,1h),4.02(m,2h),3.91(d,j=13.8hz,1h),3.78(s,3h),3.61(d,j=4.9hz,2h),3.55(d,j=17.6hz,1h),3.43(d,j=18.4hz,1h)。3.anb1h的制备步骤如下:将f5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得anb1h;1hnmr(400mhz,meod):δ7.34-7.29(m,7h),7.27-7.25(m,3h),5.64(d,j=4.8hz,1h),4.90(m,1h),4.42(d,j=13.6hz,1h),4.07(m,2h),3.98(d,j=13.6hz,1h),3.58(d,j=8.8hz,2h),3.50(d,j=7.6hz,1h),3.35(m,1h).13cnmr(101mhz,meod):δ168.03,164.57,163.42,150.82,147.28,137.43,136.53,130.87,130.65,130.42,128.08,127.12,124.44,123.17,59.01,57.80,48.05,30.11,29.45,26.12.hrms[m-h]-(m/z)forc25h22n5o4s2:calcd.520.1107,obsd.520.1138。实施例5本发明ana2c的结构式如下:1.中间产物d3的制备步骤,(1)按1:1摩尔比将g1与氨基硫脲在乙腈中混合并回流2h,冷却后所得固体用乙醇反复洗涤得中间产物g2。(2)将g2溶于氢氧化钠溶液,回流过夜后用稀盐酸调ph至酸性得到g3;1hnmr(400mhz,dmso-d6):δ13.21(s,1h),13.10(s,1h),2.74(t,j=6.1hz,2h),2.60(t,j=5.4hz,2h)。2.中间产物g4的制备步骤,(1)将a1与2,6-二甲基吡啶溶解于乙腈,0℃下滴加1.5当量的酰氯2h,然后加入等当量的三乙胺,搅拌过夜后升至室温。tlc跟踪,待反应完成后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液和饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥,抽滤,减压蒸馏除去溶剂,硅胶柱分离纯化得到a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥,减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃并在氮气保护下,缓慢加入g3,搅拌2h后升至室温继续反应。tlc跟踪,待反应完毕后除去溶剂,柱硅胶分离纯化制得g4;1hnmr(400mhz,cdcl3):δ8.02(s,1h),7.31(dd,j=7.6,5.4hz,4h),7.27-7.24(m,3h),6.98(d,j=8.9hz,1h),6.89-6.82(m,2h),5.74(dd,j=8.8,4.8hz,1h),5.22-5.10(m,2h),4.86(d,j=4.8hz,1h),4.24(d,j=13.7hz,1h),3.98(d,j=13.6hz,1h),3.78(s,3h),3.62(d,j=5.9hz,2h),3.58(s,1h),3.50-3.42(m,1h),3.00(t,j=6.7hz,2h),2.75(t,j=6.8hz,2h)。3.ana2c的制备步骤,将g4溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h。tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤获得ana2c;1hnmr(400mhz,meod):δ8.02(s,1h),7.41-7.11(m,5h),5.67(d,j=5.1hz,1h),5.01(d,j=5.1hz,1h),4.40(d,j=13.1hz,1h),4.01(d,j=13.4hz,1h),3.79(d,j=17.7hz,1h),3.65-3.54(m,2h),3.56-3.42(m,1h),2.99(m,2h),2.79(m,2h).13cnmr(101mhz,meod):δ173.97,173.26,164.74,163.40,158.47,135.09,129.37,128.94,128.87,128.23,126.67,125.42,59.21,57.66,48.11,35.66,30.34,27.23,21.29.hrms[m-h]-(m/z)forc21h20n5o6s2:calcd.502.0849obsd.502.0821。实施例6本发明anb0h结构式如下:1.中间产物h4的制备步骤,(1)将h1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体h2生成;1hnmr(400mhz,dmso-d6):δ9.81(s,1h),7.85(dt,j=7.0,1.5hz,2h),7.59-7.50(m,1h),7.38(t,j=7.4hz,2h),4.52(s,2h)。(2)将h2溶于足够量的乙醇中,室温搅拌至澄清,加入等摩尔比的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应结束后冷却至室温,过滤除去不溶物,所得滤液减压蒸馏、干燥获得h3。(3)将h3溶于氢氧化钠溶液升温至100-160℃回流,反应完毕后,用盐酸酸化至ph=5-6,所得大量白色固体用水洗至中性,真空干燥得到h4;1hnmr(400mhz,dmso-d6):δ13.5(s,1h),8.01-7.86(m,2h),7.62(dd,j=8.6,6.0hz,1h),7.55-7.46(m,2h)。2.中间产物h5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入h4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得h5;1hnmr(400mhz,cdcl3):δ7.78(d,j=9.0hz,2h),7.45-7.27(m,8h),7.33-7.22(m,2h),6.96-6.90(m,2h),6.18(d,j=9.0hz,1h),5.86(dd,j=9.1,4.9hz,1h),5.24(s,2h),4.97(d,j=4.9hz,1h),4.54(d,j=11.8hz,1h),4.44(d,j=11.8hz,1h),3.85(s,3h),3.68(d,j=8.1hz,2h),3.63(d,j=3.8hz,1h),3.46(m,1h)。3.anb0h的制备步骤如下:将h5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得anb0h;1hnmr(400mhz,meod):δ7.95(dt,j=5.8,3.6hz,2h),7.55-7.43(m,2h),7.32-7.18(m,6h),5.67(d,j=4.7hz,1h),5.04(d,j=4.7hz,1h),4.51(d,j=13.8hz,1h),4.11(d,j=13.7hz,1h),3.76(d,j=12.1hz,1h),3.64(d,j=14.7hz,1h),3.57(d,j=8.4hz,2h),3.54-3.44(m,1h).13cnmr(101mhz,meod):δ173.26,164.79,164.01,152.48,135.28,130.18,128.36,128.90,128.76,128.22,126.65,126.20,125.62,125.11,113.40,59.18,54.27,41.26,34.59,27.50.hrms[m-h]-(m/z)forc24h20n5o4s2:calcd.506.0951,obsd.506.0914。实施例7本发明anb04o结构式如下:上述anb04o制备按以下步骤进行:1.中间产物o4的制备步骤如下:(1)将o1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体o2生成;1hnmr(400mhz,dmso-d6):δ9.49(s,1h),7.97-7.42(m,2h),7.03-6.52(m,2h),4.38(s,2h)。(2)将o2溶于足够量的乙醇中,室温搅拌至澄清,加入等摩尔比的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应结束后冷却至室温,过滤除去不溶物,所得滤液减压蒸馏、干燥获得o3。(3)将o3溶于氢氧化钠溶液升温至100-160℃回流,反应完毕后,用盐酸酸化至ph=5-6,所得大量白色固体用水洗至中性,真空干燥得到o4;1hnmr(400mhz,dmso-d6):δ9.15(s,1h),7.76-7.69(m,2h),6.88-6.81(m,2h)。2.中间产物o5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入o4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得o5;1hnmr(400mhz,meod):δ7.76(d,j=8.3hz,2h),7.33-7.26(m,4h),7.26-7.22(m,1h),7.18(d,j=8.3hz,2h),6.91-6.86(m,2h),6.86-6.78(m,2h),5.66(d,j=4.9hz,1h),5.10(d,j=11.9hz,1h),5.02(d,j=4.8hz,1h),4.57(d,j=13.6hz,1h),3.95-3.88(m,1h),3.87(d,j=4.4hz,1h),3.75(s,3h),3.58(d,j=1.4hz,1h),3.55(d,j=8.4hz,1h),2.99(s,1h),2.86(s,1h)。3.anb04o的制备步骤如下:将o5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得anb04o。1hnmr(400mhz,meod):δ7.78(m,2h),7.30(m,4h),7.23(s,1h),6.92(m,2h),5.68(d,j=6.9hz,1h),5.05(d,j=6.8hz,1h),4.45(d,j=4.6hz,1h),4.15(d,j=5.0hz,1h),3.83(d,j=3.9hz,1h),3.67-3.61(m,1h),3.61-3.52(m,2h).13cnmr(101mhz,meod):δ173.27,163.49,160.08,158.86,158.42,151.32,135.08,129.04,128.86,128.46,128.22,128.06,127.65,127.38,115.64,59.17,57.68,41.78,34.50,27.24.hrms[m-h]-(m/z)forc24h20n5o5s2:calcd.522.0900,obsd.522.0923。实施例8本发明anb04n结构式如下:上述anb04n制备按以下步骤进行:1.中间产物p4的制备步骤如下,(1)将p1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体p2生成;1hnmr(400mhz,dmso-d6):δ9.49(s,1h),7.44(dd,j=8.0,1.7hz,1h),7.21-7.08(m,1h),6.71(d,j=8.2hz,1h),6.51(m,1h),6.36(s,2h),4.40(s,1h)。(2)将p2溶于足够量的乙醇中,室温搅拌至澄清,加入等摩尔比的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应结束后冷却至室温,过滤除去不溶物,所得滤液减压蒸馏、干燥获得p3。(3)将p3溶于氢氧化物钠溶液升温至100-160℃回流,反应完毕后,用盐酸酸化至ph=5-6,所得大量白色固体用水洗至中性,真空干燥得到p4;1hnmr(400mhz,dmso-d6):δ7.60-7.54(m,2h),6.72-6.65(m,2h),4.65(s,2h)。2.中间产物p5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入p4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得p5;1hnmr(400mhz,cdcl3):δ7.18(m,12h),6.87-6.73(m,2h),6.04(s,1h),5.34(dd,j=8.5,3.9hz,1h),5.21(m,1h),5.04(m,2h),4.64(s,2h),4.04(d,j=12.9hz,2h),3.99(m,1h),3.87(d,j=13.9hz,1h),3.72(s,3h),3.52(s,2h)。3.anb04n的制备步骤如下:将p5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得anb04n;1hnmr(400mhz,meod):δ7.87-7.76(m,2h),7.29(d,j=4.4hz,3h),7.23(d,j=4.5hz,2h),7.01(dt,j=8.3,3.9hz,2h),5.67(d,j=4.8hz,1h),5.03(d,j=4.7hz,1h),4.64(s,2h),4.46(d,j=13.7hz,1h),4.13(d,j=13.7hz,1h),3.86-3.75(m,1h),3.66-3.58(m,1h),3.57(d,j=8.2hz,1h),3.53-3.45(m,1h).13cnmr(101mhz,meod):δ173.01,168.95,164.37,156.70,152.74,150.25,135.09,128.95,128.27,127.83,127.20,126.70,122.37,121.08,113.48,60.89,53.15,49.65,41.80,29.68.hrms[m-h]-(m/z)forc24h21n6o4s2:calcd.521.1060,obsd.521.1051。实施例9本发明annb1h结构式如下:上述annb1h制备按以下步骤进行:1.中间产物q4的制备步骤如下:(1)将q1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体q2生成。(2)将q2溶于足够量的乙醇中,室温搅拌至澄清,将q2与氢氧化钾按1:1.5摩尔比混合,然后加入二硫化碳摩尔比(1:1.5)室温3h。过滤除去不溶物,所得滤液减压蒸馏、干燥获得q3。(3)将q3与水合联氨按1:1摩尔比混合,加热回流4h。反应完毕后,降温至室温加水,用盐酸酸化至ph=5-6,有沉淀析出,水洗真空干燥得到q4。1hnmr(400mhz,dmso-d6):δ13.54(s,1h),7.36-7.17(m,5h),5.56(s,2h),4.03(s,2h).2.中间产物q5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入q4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得q5;1hnmr(400mhz,cdcl3):δ7.19(m,12h),6.81(m,2h),6.04(s,1h),5.35(m,1h),5.21(s,1h),5.04(s,2h),4.64(s,2h),4.04(d,j=12.9hz,2h),3.99(t,j=5.3hz,1h),3.87(d,j=13.9hz,1h),3.72(s,3h),3.69-3.65(m,1h),3.61(d,j=5.3hz,2h),3.52(s,2h)。3.annb1h的制备步骤如下:将q5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得annb1h;1hnmr(400mhz,meod):δ7.34-7.27(m,10h),6.29(s,1h),5.33(d,j=6.9hz,1h),5.30(d,j=6.9hz,1h),5.20(d,j=5.3hz,1h),4.78(s,2h),4.25-4.13(m,2h),4.11-4.01(m,2h),3.76(d,j=11.9hz,1h),3.57(m,2h),3.50-3.46(m,1h).13cnmr(101mhz,meod):δ173.26,164.77,163.58,150.25,144.78,135.10,134.10,128.62,128.15,127.51,126.42,125.56,122.15,118.13,59.18,57.66,41.78,34.59,30.51,27.20.hrms[m-h]-(m/z)forc25h23n6o4s2:calcd.535.1217,obsd.535.1191。实施例10本发明anb02c的结构式如下:上述anb02c制备按以下步骤进行:1.中间产物r4的制备步骤如下:(1)将r1与水合肼按1:1摩尔比混合,升温至80℃回流8-12h。待反应完成后缓慢冷却至室温,冷却过程中有大量白色固体r2生成。(2)将r2溶于足够量的乙醇中,室温搅拌至澄清,加入等摩尔比的硫氰酸铵,然后加入浓盐酸升温至80-100℃回流12-16h。待反应结束后冷却至室温,过滤除去不溶物,所得滤液减压蒸馏、干燥获得r3。(3)将r3溶于氢氧化物钠溶液升温至100-160℃回流,反应完毕后,用盐酸酸化至ph=5-6,所得大量白色固体用水洗至中性,真空干燥得到r4;1hnmr(400mhz,cdcl3):δ8.15(dd,j=7.4,2.1hz,1h),7.82(dd,j=7.5,2.0hz,1h),7.69(td,j=7.4,2.0hz,1h),7.62(td,j=7.3,2.0hz,1h),3.28(s,1h)。2.中间产物r5的制备步骤如下:(1)将a1与2,6-二甲基吡啶溶解于乙腈并冷却至0℃,滴加1.5当量的苯乙酰氯2h,加入等当量的三乙胺,搅拌过夜后升至室温。待反应完毕后减压蒸馏除去溶剂,所得残留溶于乙酸乙酯,饱和碳酸氢钠溶液及饱和食盐水各洗涤一次,有机相用无水硫酸镁干燥、抽滤、减压蒸馏除去溶剂,硅胶柱分离纯化制得a2。(2)将a2溶于丙酮,氩气保护下按1:10摩尔比加入碘化钠,室温搅拌2.5h。反应完成后乙酸乙酯萃取,合并有机相,饱和硫代硫酸钠溶液及饱和食盐水洗涤,有机相用无水硫酸镁干燥、减压蒸馏得到a3。(3)将a3溶于氯仿,按1:1.5摩尔比加入n-甲基吗啉,0℃且在氮气保护下缓慢加入r4,0℃搅拌2h后升至室温继续反应。tlc跟踪,待反应完成后除去溶剂,柱硅胶分离纯化制得r5;1hnmr(400mhz,meod):δ7.61(m,3h),7.28(m,6h),7.23(m,2h),6.86(m,2h),5.67(d,j=5.2hz,,1h),5.14(d,j=11.9hz,1h),5.08(d,j=11.9hz,1h),5.00(d,j=4.8hz,1h),4.44(d,j=13.6hz,1h),3.97(d,j=13.6hz,1h),3.80(d,j=18.3hz,1h),3.75(s,3h),3.60-3.55(m,2h),3.55-3.51(m,1h)。3.anb02c的制备步骤如下:将r5溶于干燥的二氯甲烷,0℃下加入苯甲醚和三氟乙酸搅拌约2h,tlc监测,待反应物消耗完全,减压蒸馏除去溶剂,所得残留用乙醚反复洗涤、干燥后制得anb02c。1hnmr(400mhz,meod):δ8.00(s,1h),7.69-7.66(m,2h),7.61(m,1h),7.31-7.28(m,4h),7.24(m,2h),5.68(d,j=4.7hz,1h),5.05(d,j=4.8hz,1h),4.37(d,j=13.6hz,1h),4.17(d,j=13.7hz,1h),3.83-3.76(m,1h),3.64(d,j=5.6hz,1h),3.59(dd,j=7.7,2.4hz,2h).13cnmr(101mhz,meod)δ:173.28,168.52,164.80,161.99,158.04,151.68,135.09,132.01,130.77,130.64,130.49,129.69,128.87,128.24,127.12,126.67,120.22,113.40,59.12,53.93,42.16,34.77,27.34.hrms[m-h]-(m/z)forc25h20n5o6s2:calcd.550.0849,obsd.550.0823。实施例11药物测试主要包括以下部分:1)作为酶抑制剂,asc对mβls抑制活性的评价。2)化合物asc-na、asc-nb、anb1h、anb02c、anb04o、anb0h、anb04n、annb1h、ana2c和asc-sb,测定其协同β-内酰胺类抗生素对携带质粒pet-26-l1、pet-26-ndm-1、pet-26-vim-2、pet-26-ccra或pet-26-imis的超级耐药细菌e.coli的抑菌活性(mic)。3)以mrsa43300、普通大肠杆菌dl219(de3)为模式菌,通过抑菌效能评价初步筛选具有潜在抑菌活性或增效活性的化合物。4)asc在治疗金黄色葡萄球菌感染中的应用。5)asc-nb治疗mrsa感染的效力随其浓度和时间的依赖性变化。6)小鼠全身mrsa感染模型的建立与asc在体内治疗mrsa感染的初步评价。(一)作为酶抑制剂,asc对mβls抑制活性的评价;本实例考察包含:作为酶抑制剂,asc对mβls的广谱抑制活性评价;asc对mβls的抑制活性评价,通过测定asc对mβls的半抑制浓度(ic50)来实现。ic50的测定采用如下步骤:(1)asc对mβls的ic50值的测定在安捷伦uv8453分光光度计上进行,使用50mmtris(ph7.0)作为缓冲液。mβls选用来自三个组群并具有代表性的ndm-1、vim-2、ccra、imis和l1对asc进行评价,其最终浓度分别为13、50、58及100nm。使用50μm的头孢唑林钠作为ndm-1、vim-2、ccra和l1的底物,使用60μm亚胺培南作为imis的底物,其监测波长分别为265和300nm。(2)室温下将mβls和抑制剂在50mmtris(ph7.0)中预混合,然后将底物加入,在每个抑制剂浓度下记录抗生素水解的初始速率。所有水解速率的测定平行三次测定,取其平均值拟合得到ic50值。asc对四种mβls的抑制活性通过酶抑制动力学进行了测定,测定所得ic50值列于表1。表1asc对mβls的抑制活性(ic50,μm)从表1可知,asc对分属三个不同组群的五种mβls具有广谱抑制效能,且抑制效果良好(ic50<45μm)。其次,asc对l1具有最好的抑制活性(ic50<3.5μm)。值的指出的是,这种对“超级细菌”耐药靶蛋白mβls具有广谱抑制效能的asc,对人类攻克细菌耐药之难题带来了希望。(二)作为酶抑制剂,asc协同β-内酰胺类抗生素对携带质粒pet-26-l1、pet-26-ndm-1、pet-26-vim-2、pet-26-ccra或pet-26-imis之耐药细菌e.coli的抗菌活性评价:作为酶抑制剂,asc协同β-内酰胺类抗生素对携带质粒pet-26-l1、pet-26-ndm-1、pet-26-vim-2、pet-26-ccra或pet-26-imis之耐药细菌e.coli的抗菌活性评价,是通过测定在asc存在下抗生素对细菌最低抑制浓度(mic)的改变来实现。mic的测定,采用肉汤稀释法按如下步骤进行:(1)抗菌药物贮存液制备:制备浓度为256μg/ml的抗菌药物贮存液,贮存于-80℃超低温冰箱。选用青霉素类、头孢菌素类和碳青霉烯系列的β-内酰胺抗生素为抗菌药物。(2)培养基制备:培养基使用mueller-hinton(mh)肉汤培养基。(3)接种物的制备:选用e.coli、美国组织培养库atccmrsa43300、携带质粒pet-26-l1、pet-26-ndm-1、pet-26-vim-2、pet-26-ccra或pet-26-imis之耐药细菌e.coli为实验菌株。用接种环挑取形态相似的菌落3-5个,接种于4-5ml的mh肉汤培养基中,37℃孵育2-6小时。增菌后的对数生长期菌液用mh肉汤校正浓度至0.5麦氏比浊标准。(4)稀释抗菌药物的制备及菌液接种:取无菌管13支排序,除第1管加入1.6mlmh肉汤外,其余每管加入mh肉汤1ml,在第1管加入抗菌药物原液(128μg/ml)0.4ml混匀,然后吸取1ml至第2管,混匀后再吸取1ml至第3管,如此连续倍比稀释至第11管,并从第11管中吸取1ml弃去,第12管为不含药物的空白对照。此时各管药物浓度依次为256、128、64、32、16、8、4、2、1、0.5、0.25μg/ml。在每管内加入上述制备好的接种物各1ml,使每管菌液终浓度约为5×105cfu/ml。第1至第11管药物浓度分别为128、64、32、16、8、4、2、1、05、0.25、0.125μg/ml。测量抑制剂存在时的mic值时,取无菌试管13支排序,除第1管加入1.4mlmh肉汤外,其余每管加入mh肉汤0.9ml;第1管加入抑制剂0.2ml溶液,2-13管分别加入0.1ml,其余步骤同上。(5)孵育:接种好的稀释管塞好塞子,置37℃普通空气孵箱中孵育16-20小时。(6)结果:以肉眼或酶标仪观察,无细菌生长的试管的药物浓度,即为受试菌的mic。鉴于asc是抗生素耐药靶蛋白mβls良好的广谱型抑制剂,我们测定了asc协同先锋ⅴ对产ndm-1耐药细菌e.coli-dh10b的mic值。asc的实验浓度范围为1-64μg/ml。测定结果列于表2。表2asc协同先锋ⅴ对产ndm-1耐药细菌e.coli-dh10b的mic值(μg/ml)从表2可看出,asc自身是一种杀菌剂,其mic为128μg/ml(除annb1h外),同时,asc可有效协同抗生素提高其抗菌活性,即降低抗生素的mic值;随着asc浓度的增加,抗生素对耐药细菌的mic值降低,即其抗菌活性增强;当联用剂量64μg/ml的anb1h、anb0h、anb04n或ana2c时,使得先锋ⅴ治疗产ndm-1耐药菌e.coli的活力提高了32倍。鉴于asc是抗生素耐药靶蛋白mβls的广谱型抑制剂,asc协同先锋ⅴ对产ccra耐药细菌e.coli-bl21的mic值被测定。asc的实验浓度范围为0.5-32μg/ml。测定结果见表3。表3asc协同先锋ⅴ对产ccra耐药细菌e.coli-bl21的mic值(μg/ml)从表3可看出,asc自身是一种杀菌剂,也可有效协同抗生素提高其抗菌活性,即降低抗生素的mic值;随着asc浓度的增加,抗生素对耐药细菌的mic值降低,即其抗菌活性增强;当联用剂量8μg/ml的asc-na、anb04o或ana2c,使得先锋ⅴ治疗产ccra之耐药细菌e.coli-bl21的活力提高了32倍。(三)作为抗菌试剂,asc在治疗mrsa和大肠杆菌dl219(de3)感染中的应用:本实例考察包含:asc靶向治疗mrsa和协同抗生素治疗大肠杆菌dl219(de3)感染的效能评价;asc-nb对mrsa的杀菌效果及其浓度和时间依赖性评价;小鼠全身mrsa感染模型的建立与asc体内治疗mrsa效果的初步评价;1.以mrsa43300、大肠杆菌dl219(de3)为模式菌,通过抑菌实验初步筛选具有潜在抑菌活性或增效活性的化合物。鉴于asc为mβls良好的广谱型抑制剂,asc协同先锋ⅴ对e.coli-dl219(de3)的mic值被测定。asc的实验浓度范围为0.25-16μg/ml。测定结果见表4。表4asc协同先锋ⅴ靶向灭活e.coli-dl219(de3)的mic值(μg/ml)如表4所列数据所示,本发明所列10个新化合物asc(除annb1h外),均对e.coli-dl219(de3)具有显著地灭活效能(mic≤64μg/ml);asc协同先锋ⅴ,显著提高了该抗生素的灭菌活性。当联用剂量16μg/ml的asc-nb或aba2c,使得先锋ⅴ治疗e.coli-dl219(de3)的活力提高达64倍;当先锋ⅴ与asc-nb或aba2c的用量比为1:4时,展现出最佳协同灭菌效能。鉴于asc是抗生素耐药靶蛋白金属β-内酰胺酶的广谱型抑制剂,本发明所列asc系列10个化合物以及苯唑西林和甲氧西林(作为对照)靶向抑制mrsa的mic值被测定,其结果列于表5。表5asc靶向抑制mrsa的mic(μg/ml)化合物asc-naasc-nbanb1hanb02canb04oanb0hmic(μg/ml)324816432化合物anb04nannb1hana2casc-sb苯唑西林甲氧西林mic(μg/ml)212864888从表5可知,asc(除annb1h外)均对mrsa具有良好的抑制效果(mic≤64μg/ml),其中asc-nb、anb04o及anb04n的灭菌效力比苯唑西林及甲氧西林强2-4倍,而anb04n的灭菌效果最佳(mic=2μg/ml)。这些结果表明,asc完全有可能成为用于临床对付mrsa的潜在抗菌药物。(四)asc在治疗金黄色葡萄球菌感染中的应用:本实例考察包含:作为抗菌试剂,asc对革兰氏阴性菌和革兰氏阳性菌的抑菌活性评价;作为酶抑制剂,asc协同抗生素靶向性治疗金黄色葡萄球菌感染的效能评价。1.asc对革兰氏阴性菌和革兰氏阳性菌的抑菌活性评价:作为抗菌试剂,asc的抑菌活性评价是通过测定其对细菌的最低抑制浓度(mic)来实现(mic的测定详见下)。分别以革兰氏阳性菌和革兰氏阴性菌为实验菌株,以asc作为抗菌试剂测定的mic值列于表6:表6asc的mic值(μg/ml)附注:“-”表示在128μg/ml剂量的asc存在时,实验菌株仍有活性。从表6可知,在5个革兰氏阳性菌和革兰氏阴性菌为实验菌株当中,asc仅仅对金黄色葡萄球菌展现出灭菌活性,且灭菌活性非常的好(mic<1μg/ml)。这一评价揭示,asc对金黄色葡萄球菌具有专一性灭活作用。2.asc协同抗生素靶向性治疗金黄色葡萄球菌感染的效能评价:作为酶抑制剂,asc协同抗生素靶向性治疗金黄色葡萄球菌感染的效能评价,是通过测定在asc存在下抗生素对细菌最低抑制浓度(mic)的改变来实现。mic的测定,采用肉汤稀释法按如下步骤进行:(1)抗菌药物贮存液制备:制备浓度为128μg/ml的抗菌药物贮存液,贮存于-80℃超低温冰箱。选用青霉素、头孢菌素和碳青霉烯系列的β-内酰胺抗生素做为抗菌药物;(2)培养基制备:培养基使用mueller-hinton(mh)肉汤培养基;(3)接种物的制备:选用e.coli、临床上分离到的esbl-e.coli、铜绿假单胞菌、粪肠杆菌、标准的esbl-肺炎克雷伯菌(atcc700603)和金黄色葡萄球菌为实验菌株。用接种环挑取形态相似的菌落3-5个,接种于4-5ml的mh肉汤培养基中,35℃孵育2-6小时。增菌后的对数生长期菌液用mh肉汤校正浓度至0.5麦氏比浊标准;(4)稀释抗菌药物的制备及菌液接种:取无菌管13支排序,除第1管加入1.6mlmh肉汤外,其余每管加入mh肉汤1ml,在第1管加入抗菌药物原液(128μg/ml)0.4ml混匀,然后吸取1ml至第2管,混匀后再吸取1ml至第3管,如此连续倍比稀释至第11管,并从第11管中吸取1ml弃去,第12管为不含药物的空白对照。此时各管药物浓度依次为256、128、64、32、16、8、4、2、1、0.5、0.25μg/ml。在每管内加入上述制备好的接种物各1ml,使每管菌液终浓度约为5×105cfu/ml。第1至第11管药物浓度分别为128、64、32、16、8、4、2、1、05、0.25、0.125μg/ml。测量抑制剂存在时的mic值时,取无菌试管13支排序,除第1管加入1.4mlmh肉汤外,其余每管加入mh肉汤0.9ml;第1管加入抑制剂0.2ml溶液,2-13管分别加入0.1ml,其余步骤同上;(5)孵育:接种好的稀释管塞好塞子,置35℃普通空气孵箱中孵育16-20小时;(6)结果:以肉眼或酶标仪观察,无细菌生长的试管的药物浓度,即为受试菌的mic。鉴于asc是抗生素耐药靶蛋白金属β-内酰胺酶良好的广谱型抑制剂,我们评价了asc协同抗生素靶向性治疗金黄色葡萄球菌感染的效能。评价实验选取了临床上日常使用的三大类8种抗生素,asc的实验浓度范围为0.06125-1μg/ml。测定的不同浓度实施例1化合物asc-nb和和实施例3化合物asc-sb协同不同种类抗生素靶向抑制金黄色葡萄球菌的mic分别列于表7和8。表7asc-nb协同不同种类抗生素对金黄色葡萄球菌的灭菌活性(mic,μg/ml)如表7所示,除卡那霉素外,asc-nb可协同β-内酰胺类和四环素类抗生素共7种实验的抗生素(青霉素g、氨苄西林、头孢唑林钠、头孢曲松钠、头孢噻肟钠、亚胺培南、四环素),展现出对金黄色葡萄球菌的剂量依赖性灭菌活性,即随着asc-nb剂量的增加,抗生素对金葡菌的灭菌活性在增强(mic值降低)。当联用1μg/ml剂量的asc-nb,上述抗生素对金葡菌的灭菌能力分别提高了64、128、4、32、16、2、8倍以上。asc-nb与氨苄西林展现出最佳协同效能,即联用1μg/ml剂量的asc-nb,使用目前临床上用量1/128剂量的氨苄西林就可治疗金葡菌的感染。表8asc-sb协同不同种类抗生素对金黄色葡萄球菌的灭菌活性(mic,μg/ml)由表8可知,asc-sb可协同β-内酰胺类、氨基糖苷类和四环素类抗生素共8种实验的抗生素(青霉素g、氨苄西林、头孢唑林钠、头孢曲松钠、头孢噻肟钠、亚胺培南、卡那霉素、四环素),展现出对金黄色葡萄球菌的剂量依赖性灭菌活性,即随着asc-sb剂量的增加,抗生素对金葡菌的灭菌活性在增强(mic值降低)。当联用1μg/ml剂量的asc-sb,上述抗生素对金葡菌的灭菌能力分别提高了64、128、4、32、16、10、16、4倍以上。同样,asc-sb与氨苄西林展现出最佳协同效能,即联用1μg/ml剂量的asc-nb,使用目前临床上用量1/128剂量的氨苄西林既可治疗金葡菌的感染。(五)asc灭活mrsa和金黄色葡萄球菌的剂量及时效依赖性评价:作为一类新型抗菌药物,我们选择asc-na、asc-nb(用先锋ⅴ作为对照)评价了其灭活mrsa或金黄色葡萄球菌的剂量及时间依赖性。将不同浓度的asc-na、asc-nb(先锋ⅴ)溶液加入至等体积等密度(od600=0.5)的mrsa或金黄色葡萄球菌菌液中,在不同时间、不同给药量时测定实验菌的od600值。测定结果见图1~3所示。图1清楚地显示,asc-na对金黄色葡萄球菌展现出剂量依赖性灭活效能(这与前述mic测定结果是一致的),但使用一个剂量为2μg/ml的asc-na,金黄色葡萄球菌的感染几乎100%地被抑制,其灭菌效能维持至少48小时以上。图2清楚显示,先锋ⅴ对mrsa展现出剂量依赖性灭活性能(与前述mic测定结果一致)。但是,当使用一个剂量为16μg/ml的先锋ⅴ,当时间达到48h时,mrsa的感染并没有100%地被灭活,并再次产生了一定的耐药性。对照图2看图3可知,asc-nb对mrsa展现出剂量依赖性灭活效能(这与前述mic测定结果一致),即随着给药量的逐渐提高,耐药细菌的密度逐渐降低;当使用一个剂量仅为16μg/ml的asc-nb,便可将mrsa几乎100%地灭活,其灭菌效能维持至少48小时以上。显然,asc是一类良好的抗mrsa药物。(六)小鼠全身感染mrsa模型的建立与asc体内治疗mrsa效果的初步评价:建立稳定、可靠的mrsa全身感染模型,以对系统地研究mrsa感染的发病机制以及治疗奠定实验基础。采用国际标准菌株mrsa43300建立od600-cfu标准曲线,经腹腔注射途径感染昆明小鼠,从感染剂量的选择、小鼠的生存率、小鼠体重变化、血液及多脏器细菌定殖量等层面进行时相性监测,对建立的小鼠模型进行系统评价。经此途径建立的小鼠模型,致死剂量为每只5.0×109cfu,亚致死剂量为每只1.0×109cfu(生存率为60%-70%)。材料方法如下:(1)实验动物:昆明小鼠,雄雌各半,6-8周龄,体重18-22g,购自西安交通大学医学院动物实验中心。(2)菌株:国际标准菌株mrsa43300,购自美国组织培养库(americantissueculturecollection,atcc)。(3)感染用菌株的培养与制备:复苏国际标准菌株mrsa43300,划线接种于mh平板,37℃,适当转速,培养6-8h,离心后弃上清,所得菌落再以无菌生理盐水洗涤两次,最后以无菌生理盐水重悬菌体,测定其在600nm处的吸光度(a600)。根据以往实验所得a600-cfu标准曲线,调整菌液浓度约为5.0×109cfu/ml。(4)小鼠模型的建立:将小鼠随机分为4组,每组10只。其中实验组3组,对照组1组。所有小鼠提供足够的饲料和饮水。实验组小鼠(第ⅰ、ⅱ、ⅲ组)分别经腹腔注射0.2ml重悬在生理盐水中的菌体,对照组小鼠经腹腔注射等量无菌生理盐水。4h后,将剂量为35mg/kg的asc-nb或苯唑西林钠分别腹腔注射到第ⅱ、ⅲ实验组小鼠体内,对照组和实验组(i)不给药。治疗后,将小鼠解剖,取其五脏研磨成匀浆并进行涂布于培养皿,37℃繁殖过夜后计菌落数。结果如图4所示。从图4可以看出,mrsa感染组小鼠的心、肝、脾、肺和肾的细菌定殖量达到最大,使用剂量为35mg/kg的苯唑西林治疗感染mrsa的小鼠,小鼠各器官组织除脾外的细菌定殖量明显降低,即苯唑西林对mrsa有一定疗效;然而,使用同剂量的asc-nb,小鼠五脏中的细菌定殖量比用苯唑西林更为显著降低,特别是心脏和肺部感染的mrsa几乎全部被灭杀,且维持一周没有观察到mrsa长出。显而易见,asc-nb对mrsa有疗效,其疗效比苯唑西林更好,特别是对心脏和肺部感染的mrsa的治疗,具有用于临床治疗mrsa感染的潜在应用价值。综上,金黄色葡萄球菌和mrsa是目前临床上最重要的也是最难处理的致病菌。基于20年来我们对细菌耐药性的研究,发现asc是一类抗生素耐药靶蛋白金属β-内酰胺酶(mβls)良好的广谱型抑制剂。作为一类新型抗菌药物,asc重要的临床价值在于:asc可协同7-8种抗生素(青霉素g、氨苄西林、头孢唑林钠、头孢曲松钠、头孢噻肟钠、亚胺培南、卡那霉素、四环素等)靶向性治疗金黄色葡萄球菌的感染。联用1μg/ml剂量的asc,使得这些抗生素的活力提高4-128倍以上。即联用1μg/ml剂量的asc,使用目前临床上剂量1/128的氨苄西林就可灭活金黄色葡萄球菌;一个2μg/ml剂量的asc-na几乎100%地治疗金黄色葡萄球菌的感染,其效能维持至少48小时以上。此外,asc可有效治疗mrsa的感染,其中anb04n的治疗效果最佳(mic=2μg/ml),要比苯唑西林灭活mrsa的效力强达4倍。asc-nb对mrsa展现剂量依赖性灭活性能,一个剂量为16μg/ml的asc-nb几乎100%地灭活mrsa,其灭菌效能维持48小时以上。动物(小鼠)实验表明,asc-nb极有效地治疗小鼠感染的mrsa。一个给药量为35mg/kg的asc-nb,使得小鼠心脏和肺部感染的mrsa几乎全部被杀死,一周后仍无细菌繁殖;同剂量的asc-nb,也使得小鼠的肝、脾和肾组织中mrsa的定殖量显著降低。asc-nb治疗mrsa的效能随给药浓度和用药时间的不同而呈依赖性变化。显然,asc-nb对小鼠感染的mrsa有很好的治疗效果,这预示:asc具有成为临床治疗mrsa感染的潜在药物的可能性。以上内容是结合具体的优选实施方式对本发明所做出的进一步详细说明,不能认定是唯一的实施方式。本领域普通技术人员通过阅读本发明说明书而对本发明技术方案采取的任何等效的变换,均应视为本发明的权利要求书所涵盖。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1