一种金属镁配合物、制备方法及其应用与流程
本发明涉及金属镁配合物技术领域,且特别涉及一种金属镁配合物、制备方法及其应用。
背景技术:
金属有机配合物因具有新颖的结构、有趣的磁性、光学等性质,其设计、合成已经成为无机化学研究的一个热点。目前,金属有机配合物已被广泛应用在了感应器件、气体储存、分子吸收以及分离、离子交换、催化等领域。在金属有机配合物的制备过程中,配体的构型对配合物的最终结构起着至关重要的作用。
金属镁配合物的设计合成与研究丰富了金属配位化学的研究,有助于获得有益的发光性能荧光材料。其中,新颖金属有机配合物的构筑是其研究的一个热点,选择合适的配体设计配合物自组装条件为碱土金属配合物的构筑提供了可行的策略。
技术实现要素:
本发明的目的在于提供一种金属镁配合物、制备方法及其应用,该金属镁配合物是一种新的金属镁配合物,首次发现并合成,具有优异的性质。
本发明解决其技术问题是采用以下技术方案来实现的。
本发明提供一种金属镁配合物,该金属镁配合物的化学式为mg1.5(l)1·(c2h7n)·(h2o),其中,l为均四苯甲酸。
本发明还提供一种上述金属镁配合物的制备方法,包括以下步骤:将六水合硝酸镁、均苯四甲酸和吡嗪溶解、加热,制得金属镁配合物。
本发明还提供一种上述金属镁配合物的应用,将金属镁配合物用于荧光识别或分子检测领域。
本发明的有益效果是:
本发明提供了一种金属镁配合物、制备方法及其应用,本发明中的配合物选择苯羧酸类为有机配体,引入合适的配体可以实现配合物的结构调控、性质改变等,获得具有独特结构和性质的碱土金属配合物。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
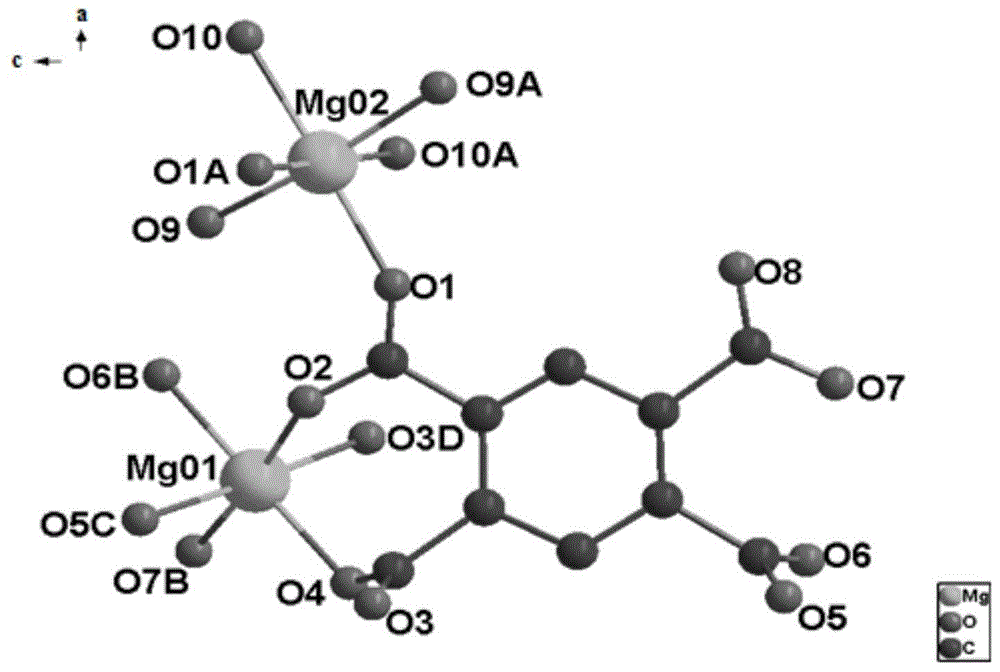
图1为本发明实施例1-4中的金属镁配合物的分子结构示意图;
图2为本发明实施例1-4中的金属镁配合物的x射线衍射图;
图3为本发明实施例1-4中的金属镁配合物的荧光发射光谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
下面对本发明实施例提供的一种金属镁配合物、制备方法及其应用进行具体说明。
本发明实施例提供一种金属镁配合物,该金属镁配合物的化学式为mg1.5(l)1·(c2h7n)·(h2o),其中,l为均四苯甲酸。
本发明实施例提供一种金属镁配合物,该金属镁配合物的化学式为mg1.5(l)1·(c2h7n)·(h2o),其中,l为均四苯甲酸。该金属配合物选择镁作为金属,选择苯羧酸类为有机配体,引入合适的辅助配体可以实现配合物的结构调控、性质改变等一系列目的,能够获得具有独特结构和性质的碱土金属配合物。
金属镁配合物的设计合成与研究丰富了金属配位化学的研究,有助于获得有益的发光性能荧光材料。而羧酸类配体具有丰富的配位点和多样的配位方式,可以与碱土金属镁原子形成较强的配位键,从而构筑出具有新颖结构金属镁配合物。其次,羧酸类有机配体的共轭效应有助于制备新型发光材料,利用该类配体与碱土金属镁构筑的荧光材料有助于理解配合物中碱土金属离子与配体之间的相互作用,在荧光识别和分子检测等方面具有潜在的应用。
在一些实施方式中,金属镁配合物属于斜方晶系,空间群为pbcn,其晶胞参数为:α=90°,β=90°,γ=90°。
本发明实施例还提供一种上述金属镁配合物的制备方法,包括以下步骤:将六水合硝酸镁、均苯四甲酸和吡嗪溶解、加热,制得金属镁配合物。
本发明实施例提供一种上述金属镁配合物的制备方法,优选的,镁源为六水合硝酸镁,羧酸配体来源为均苯四甲酸,原料六水合硝酸镁、均苯四甲酸和吡嗪配位生成金属镁配合物,吡嗪通过改善六水合硝酸镁、均苯四甲酸的配位时的酸碱性或配位环境,来提高原料的配合程度,若镁源为氧化镁或氯化镁或硫酸镁等其他的镁盐,由于上述的镁盐可能参与配合物的生长,导致产生的晶体的结构发生变化,纯度降低。
在一些实施方式中,六水合硝酸镁的用量为0.95-1.10wt%,均苯四甲酸的用量为0.65-0.90wt%,吡嗪的用量为0.20-0.35wt%。
在一些实施方式中,溶解时所用的溶剂包括n,n-二甲基甲酰胺、乙腈以及水。
在一些实施方式中,溶剂的用量为:n,n-二甲基甲酰胺50-55wt%、乙腈28-33wt%以及水12.5-15.5wt%。
在一些实施方式中,溶解为密闭溶解,优选的,溶解为超声震荡溶解,且超声震荡的时间为5-10min。
在一些实施方式中,加热的温度为90-105℃,时间为64-90小时。
本发明实施例中的配合物的制备过程中的加热的温度为90-105℃,时间为64-90小时。在上述的温度和时间下,六水合硝酸镁、均苯四甲酸和吡嗪配位生成金属镁配合物,生成结构如图1所示的配合物,如果温度低于或者高于上述的温度,会导致无法生成所示结构的配合物或者配合物的结构发生改变。
在一些实施方式中,还包括:加热结束后冷却至室温,用乙腈清洗2-3次,过滤,得到白色针状晶体。
本发明实施例还提供一种上述金属镁配合物的应用,将金属镁配合物用于荧光识别或分子检测领域。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1
一种金属镁配合物mg1.5(l)1·(c2h7n)·(h2o)的制备方法,包括以下步骤:
取一10毫升玻璃瓶,将硝酸镁(mg(no3)2·6h2o)(1.05wt%),均苯四甲酸(c10h2o8)(0.7wt%),吡嗪(c4h4n2)(0.33wt%)加入瓶中。
将n,n-二甲基甲酰胺(dmf)溶剂(51.87wt%),乙腈(c2h3n)(32.40wt%),水(h2o)(13.65wt%)加入瓶中。
将玻璃瓶封口密闭后超声震荡5-7分钟(震荡频率为100w)。
震荡后在恒温箱中进行加热,温度为90-100℃,时间72-80小时。
加热结束后冷却至室温,然后采用乙腈进行清洗3次,经过滤后即得白色针状mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物晶体。
实施例2
一种金属镁配合物(mg1.5(l)1·(c2h7n)·(h2o))的制备方法,包括以下步骤:
取一10毫升玻璃瓶,将硝酸镁(mg(no3)2·6h2o)(0.96wt%),均苯四甲酸(c10h2o8)(0.65wt%),吡嗪(c4h4n2)(0.20wt%)加入瓶中。
将n,n-二甲基甲酰胺(dmf)溶剂(51.19wt%),乙腈(c2h3n)(33wt%),水(h2o)(14wt%)加入瓶中。
将玻璃瓶封口密闭后超声震荡7-10分钟(震荡频率为100w)。
震荡后在恒温箱中进行加热,温度为100-105℃,时间64-72小时。
加热结束后冷却至室温,然后采用乙腈进行清洗3次,经过滤后即得白色针状mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物晶体。
实施例3
一种金属镁配合物(mg1.5(l)1·(c2h7n)·(h2o))的制备方法,包括以下步骤:
取一10毫升玻璃瓶,将硝酸镁(mg(no3)2·6h2o)(1wt%),均苯四甲酸(c10h2o8)(0.67wt%),吡嗪(c4h4n2)(0.30wt%)加入瓶中。
将n,n-二甲基甲酰胺(dmf)溶剂(55wt%),乙腈(c2h3n)(28.03wt%),水(h2o)(15wt%)加入瓶中。
将玻璃瓶封口密闭后超声震荡6-9分钟(震荡频率为100w)。
震荡后在恒温箱中进行加热,温度为95-100℃,时间80-90小时。
加热结束后冷却至室温,然后采用乙腈进行清洗3次,经过滤后即得白色针状mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物晶体。
实施例4
一种金属镁配合物(mg1.5(l)1·(c2h7n)·(h2o))的制备方法,包括以下步骤:
取一10毫升玻璃瓶,将硝酸镁(mg(no3)2·6h2o)(1.1wt%),均苯四甲酸(c10h2o8)(0.8wt%),吡嗪(c4h4n2)(0.35wt%)加入瓶中。
将n,n-二甲基甲酰胺(dmf)溶剂(51.75wt%),乙腈(c2h3n)(33wt%),水(h2o)(13wt%)加入瓶中。
将玻璃瓶封口密闭后超声震荡5-9分钟(震荡频率为100w)。
震荡后在恒温箱中进行加热,温度为95-100℃,时间72-84小时。
加热结束后冷却至室温,然后采用乙腈进行清洗3次,经过滤后即得白色针状mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物晶体。
实施例5
测试结果
参见图1,本发明实施例1-4制备的mg金属配合物呈三维结构,属于斜方晶系,pbcn空间群,晶体的最小不对称单元中,两种位置的mg均为六配位。mg01上,羧酸上的o3以单齿的配位方式与mg配位,羧酸上的o5以反-反双齿桥联的配位方式与mg配位,羧酸上的o2、o4是同一个均苯四甲酸上3、4位上的羧基,以反-反双齿桥联的方式与mg配位,且与同一个mg进行配位。o6与o7是同一个均苯四甲酸上的两个羧基以单齿配位桥联的方式与mg配位。mg02同样也是6配位,其与6个o进行配位,但是只有两个o在结构中起着桥梁的作用,均苯四甲酸中的一个羧基以单齿配位方式和mg配位,同一均苯四甲酸中3、4位上的两个羧基连接两个mg,也有同一均苯四甲酸上的两个羧基桥联着3个mg。
参见图2,本发明实施例1-4制备的mg金属配合物的x射线衍射图。
由图2可以看出:本发明实施例1-4中的制备的mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物结构稳定,结晶度良好。其中,x射线衍射图中的衍射峰强度高,本底小,各个峰尖锐,衍射峰的晶面间距和相对强度与粉末衍射卡中的六水合硝酸镁、均苯四甲酸和吡嗪完全不同,表明得到的产物中原料全部作用生成新的化合物,而不是六水合硝酸镁、均苯四甲酸的混合。六水合硝酸镁、均苯四甲酸和吡嗪的衍射峰完全消失,说明新的晶体配合物已生成,我们采用wernen的面指数尝试法计算机程序对x射线衍射数据进行指标化标定,结果表明,所有衍射峰均可用一套晶胞参数进行拟合,所计算的晶面间距与实测值非常接近,可以看出合成的金属镁配合物具有很好的纯度。确定出该配合物为属于斜方晶系,空间群为pbcn,其晶胞参数为:α=90°,β=90°,γ=90°,具体的晶胞参数如下:
图3为本发明的实施例1-4提供的mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物的荧光发射光谱图。
由图3可以看出:本发明实施例1-4中的制备的mg1.5(l)1·(c2h7n)·(h2o)镁金属配合物是一种发光性能荧光材料,其最大发射波长约为410nm。
综上,本发明实施例提供了一种金属镁配合物、制备方法及其应用,该金属镁配合物的化学式为mg1.5(l)1·(c2h7n)·(h2o),其中,l为均四苯甲酸。该金属镁配合物选择苯羧酸类为有机配体,引入合适的辅助配体可以实现配合物的结构调控、性质改变等一系列目的,能够获得具有独特结构和性质的碱土金属配合物。
以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!