一种西他沙星有关物质D-3的制备方法与流程
本发明属于有机合成技术领域,具体是涉及一种西他沙星有关物质d-3的制备方法。
背景技术:
西他沙星(sitafloxacin),化学名为(-)-7-[(7s)-7-氨基-5-氮杂螺[2.4]庚-5-基]-8-氯-6-氟-1-[(1r,2s)-2-氟-1-环丙基]-1,4-二氢-4-氧代-3-喹啉羧酸,是日本第一制药三共株式会社开发的广谱喹诺酮类抗菌药,2008年首次在日本上市。临床用其3/2水合物,用于治疗严重难治性细菌感染、复发性感染以及某些耐药菌感染。西他沙星能抑制细菌dna促旋酶和拓扑同工酶,对该两种酶的抑制作用较其它喹诺酮类药物要强。
西他沙星结构式如式i所示:
西他沙星最终产品中,有关物质d-3(化合物ii)化学名为8-氯-6-氟-1-[(1r,2s)-2-氟-1-环丙基]-7-羟基-1,4-二氢-4-氧代-3-喹啉羧酸,是西他沙星酸if文件中提到的降解杂质中的一种,其结构式如下所示:
目前还没有文献公开报道西他沙星有关物质d-3(化合物ii)的合成方法。
技术实现要素:
本发明的目的是提供一种西他沙星有关物质d-3的制备方法,为西他沙星的质量控制提供合格的对照品。
本发明是通过以西他沙星中间体iii为起始原料,经过环合、酸性水解或者直接采用中间体v为原料,由中间体v直接酸性水解得到反应液,最后经过后处理得到西他沙星有关物质d-3。
本发明的反应路线如下:
一种西他沙星有关物质d-3的制备方法,包括:化合物(v)在混酸中,在90℃以上,反应20小时以上,后处理得到西他沙星有关物质d-3;
所述化合物(v)结构如下式所示:
所述西他沙星有关物质d-3结构如下式所示:
作为优选,所述后处理方法包括:
(i-1)向得到的反应液中加入水和二氯甲烷,收集水相,浓缩得到西他沙星有关物质d-3粗品;
(i-2)将得到的西他沙星有关物质d-3粗品,用不溶盐或微溶盐的强极性有机溶剂升温打浆,固液分离,合并液相,浓缩得到所述西他沙星有关物质d-3。
生成的杂质大部分是西他沙星制备路线中的水解产物。利用二氯甲烷去除大部分有机杂质和未反应完全的原料。
通过上述后处理方法,可以直接得到高纯度的西他沙星有关物质d-3,不需要复杂的柱层析或者其他不适于工业化的操作,为本发明的技术方案的工业化提供前提保证。
上述后处理方法中,作为优选,加入的水的体积为混酸体积的3~10倍,这样能够保证几乎所有的产品能够被收集,同时利用二氯甲烷,去除未反应的原料以及其他杂质。
本发明中,可以直接采用化合物(v)纯品进行反应,可以购自市售产品,也可以由现有的制备方法制备得到。
当采用现有的方法制备化合物(v)时,作为优选,可以采用化合物(v)的浓缩液作为原料进行本发明的有关物质d-3的制备。作为优选,所述化合物(v)为含有化合物(v)的浓缩液,由下述方法得到:采用化合物(iii)为原料,在非质子化溶剂中,强碱存在下,进行环合反应,反应完成后,得到化合物(v)的反应液,利用稀酸淬灭反应,浓缩,得到化合物(v)的浓缩液;
所述化合物(iii)的结构如下:
采用化合物(iii)为原料时,优选的方案包括:
一种西他沙星有关物质d-3的制备方法,包括以下步骤:
(1)起始原料西他沙星中间体(iii)(即化合物(iii))在一种非质子化溶剂加入一种强碱,进行环合反应得到中间体v(即化合物(v))的反应液;
(2)上述反应液用稀酸,调节ph≤3.0,减压浓干,加入混酸溶解,高温下反应得到含有西他沙星有关物质d-3的反应液;
(3)上述反应液中加入大量水,固体析出,加入二氯甲烷溶解,静置,分层,收集水相,减压浓干得到含大量盐和西他沙星有关物质d-3的黄色固体;
(4)上述黄色固体用不溶盐或微溶盐的强极性有机溶剂升温打浆,过滤,减压浓干得到西他沙星有关物质d-3黄色固体。
本发明中,反应所述非质子溶剂包括乙腈、四氢呋喃、二氧六环中的一种或多种。
本发明中,反应所述强碱包括氢化钠、氢氧化锂、氢氧化钠、氢氧化钾、叔丁醇钾、乙醇钠和氢氧化钙中的一种或多种。
本发明中,所述环合反应温度为15~20℃。
本发明中,所述稀酸包括盐酸、硫酸、醋酸中的一种或多种。
本发明中,所述混酸(体积比)为浓盐酸:乙酸=1:1.
本发明中,所述高温温度为90~100℃。
本发明中,所述反应时间为24~30h。
本发明中,所述减压浓缩温度一般为40~50℃。
本发明中,所述强极性有机溶剂包括甲醇、乙醇、乙腈、丙酮中的一种或多种。
本发明中,所述打浆温度为50~90℃,实际操作时,作为优选,所述打浆温度一般为所使用溶剂的沸点温度。
由于目前还没有文献公开报道西他沙星有关物质d-3的合成方法,直接采用西他沙星酸降解条件制备西他沙星有关物质d-3,分离难度较大。与现有技术相比,本发明的合成方法,合成路线较为简捷,反应条件较为温和,采用原料和试剂价格便宜易得到。而且相对于一般杂质制备采用柱层析分离技术,本方法可以直接得到较多的目标产物。
附图说明
图1为实施例1制备得到的西他沙星有关物质d-3的hplc图。
图2为实施例1制备得到的西他沙星有关物质d-3的ms图。
图3为实施例1制备得到的西他沙星有关物质d-3的1hnmr图。
具体实施方式
下面通过具体实施例对发明作进一步说明。
实施例1:
于250ml四口瓶中加入20g西他沙星中间体(iii)、200ml二氧六环,搅拌溶解,降温至15-20℃,分批缓慢加入0.4g60%nah,搅拌0.5h,加入25ml乙酸,调节ph≤3.0,40-50℃减压浓干,得到中间体(v)的浓缩液,向反应瓶中加入90ml浓盐酸、90ml乙酸,搅拌,溶解,升温至95-100℃,反应24h,降至室温,滴加至1000ml水中,固体析出,加入250mldcm溶解,搅拌,静置,分层,分出水层,加入200ml×3dcm洗涤水层,收集水相,40-50℃减压浓干,加入200ml乙腈,升温至回流,搅拌1h,过滤,滤液转移至干净的反应瓶中,重复打浆操作6次。合并所有乙腈滤液,40-50℃减压浓干即可得到黄色固体西他沙星有关物质d-3,重量3.07g,纯度为97.5%(λ=295nm),见图1,收率为17.8%。
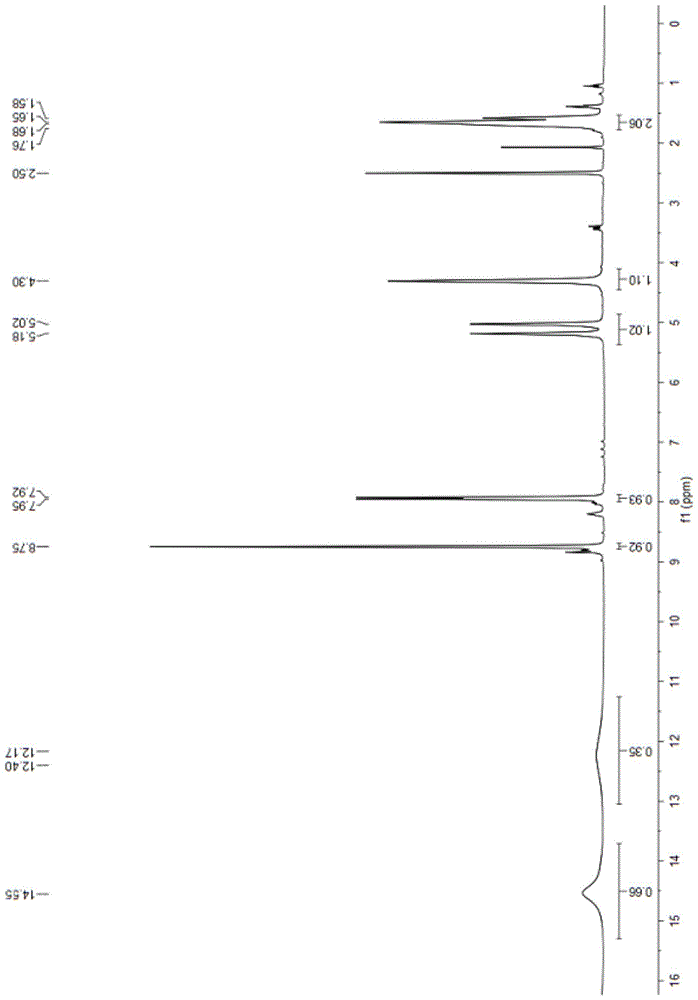
得到的西他沙星有关物质d-3的结构检测数据如下:
图2:ms(m+h)=316.0172;
图3:
1hnmr(400mhz,dmso)δ14.55(s,1h),12.29(d,j=92.7hz,1h),8.75(s,1h),7.94(d,j=9.8hz,1h),5.10(d,j=63.9hz,1h),4.30(s,1h),1.77–1.53(m,2h).
实施例2:
于250ml四口瓶中加入20g西他沙星中间体(iii)、200ml二氧六环,搅拌溶解,降温至15-20℃,分批缓慢加入0.4g60%nah,搅拌0.5h,加入25ml乙酸,调节ph≤3.0,40-50℃减压浓干,得到中间体(v)的浓缩液,向反应瓶中加入90ml浓盐酸、90ml乙酸,搅拌,溶解,升温至95-100℃,反应30h,降至室温,滴加至1000ml水中,固体析出,加入250mldcm溶解,搅拌,静置,分层,分出水层,加入200ml×3dcm洗涤水层,收集水相,40-50℃减压浓干,加入200ml乙腈,升温至回流,搅拌1h,过滤,滤液转移至干净的反应瓶中,重复打浆操作6次。合并所有乙腈滤液,40-50℃减压浓干即可得到黄色固体西他沙星有关物质d-3,重量4.21g,纯度为97.6%(λ=295nm),收率为24.4%。
实施例3
于250ml四口瓶中加入20g西他沙星中间体(iii)、200ml二氧六环,搅拌溶解,降温至15-20℃,分批缓慢加入0.4g60%nah,搅拌0.5h,加入25ml乙酸,调节ph≤3.0,40-50℃减压浓干,得到中间体(v)的浓缩液,向反应瓶中加入90ml浓盐酸、90ml乙酸,搅拌,溶解,升温至120℃~130℃,反应25h,降至室温,滴加至1000ml水中,固体析出,加入250mldcm溶解,搅拌,静置,分层,分出水层,加入200ml×3dcm洗涤水层,收集水相,40-50℃减压浓干,加入200ml乙腈,升温至回流,搅拌1h,过滤,滤液转移至干净的反应瓶中,重复打浆操作6次。合并所有乙腈滤液,40-50℃减压浓干即可得到黄色固体西他沙星有关物质d-3,重量3.25g,纯度为97.3%(λ=295nm),收率为18.8%。
实施例4
将实施例1中的钠氢替换为等摩尔量的氢氧化钠,最后得到的西他沙星有关物质d-3的纯度为97.6%,收率为28%。
对比例1
采用文献(西他沙星的合成及表征;杨桂玲,吴小明;安徽化工:第42卷,第1期)p55:1.2.3的方法:
将5.4g化合物3(即本发明中的化合物(v))、50ml浓盐酸、50ml醋酸加入反应瓶中,搅拌升温到120℃~130℃,反应2h。采用本发明实施例1的同样的后处理方法,几乎得不到本发明所要的产品西他沙星有关物质d-3。由此可知,反应时间对于本发明的产品的生成,具有关键性作用。
- 还没有人留言评论。精彩留言会获得点赞!