一种制备多取代烯基脒的方法与流程
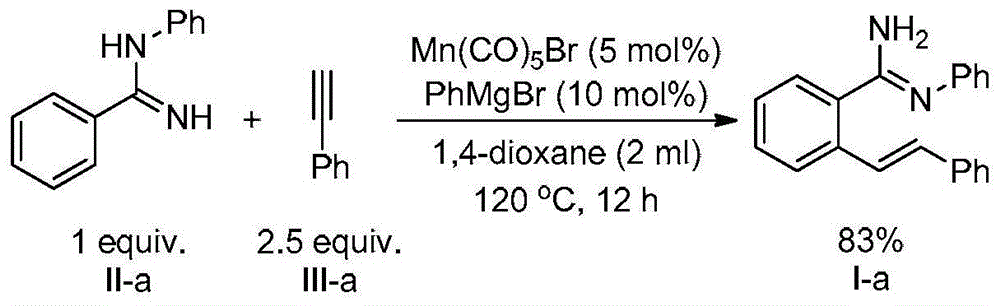
本发明属于化学合成领域,具体涉及一种制备多取代烯基脒的方法。
背景技术:
多取代烯基脒是有机化合物中重要的结构单元,在有机合成领域扮演者非常重要的角色,在医药、化工等领域有着广泛的应用。迄今为止,有关邻位多取代烯基脒的直接制备方法还从未被报道。目前邻位多取代烯基脒的合成,需从邻位氰基取代的1,2-二取代乙烯,或其相关化合物制备,但是邻氰基-1,2-二取代乙烯的制备方法尚不完善,限制了原料的多样性。为解决目前在多取代烯基脒合成中所面临的的步骤繁琐,原料昂贵等问题,发展一类方法简单、高效、价格低廉的合成手段具有重要的意义。
技术实现要素:
本发明的目的是提供一种制备多取代烯基脒的方法。
所述多取代烯基脒,其结构式如式i所示:
上述式i中,r1表示苯环上的单取代基或多取代基,为:取代或未取代的碳原子数为1~10的烷基(具体可为取代或未取代的碳原子数为1~6的烷基)、取代或未取代的碳原子数为1~10的烷氧基(具体可为取代或未取代的碳原子数为1~6的烷氧基)、芳基、卤素和与苯环稠合得到的芳环的任意一种或多种;
r2为:氢或碳原子数为1~10的烷基(具体可为碳原子数为1~6的烷基);
r3为:芳基、杂芳基或苯环稠合得到的芳环中的任意一种。
具体地,r1为氢、甲基、甲氧基、三氟甲基、氟、氯、溴、苯基或与苯环稠合得到的芳环;
r2为氢或甲基;
r3为苯基,含杂原子取代基的苯基(如卤素取代的苯基),对甲基苯基,对甲氧基苯基或杂芳基。
所述制备多取代烯基脒的方法,包括如下步骤:
在锰催化剂和格氏试剂存在下,使得式ii所示脒与式iii所示炔烃经c-h活化/c-c形成反应,得到式i所示多取代烯基脒;
上述式ii中,r1表示苯环上的单取代基或多取代基,为:取代或未取代的碳原子数为1~10的烷基(具体可为取代或未取代的碳原子数为1~6的烷基)、取代或未取代的碳原子数为1~10的烷氧基(具体可为取代或未取代的碳原子数为1~6的烷氧基)、芳基、卤素或与苯环稠合得到的芳环的任意一种或多种;
具体地,r1为氢、甲基、甲氧基、三氟甲基、氟、氯、溴、苯基或与苯环稠合得到的芳环;
上述式iii中,r2为:氢或碳原子数为1~10的烷基(具体可为碳原子数为1~6的烷基);具体地,r2为氢或甲基;
r3为:芳基、杂芳基或苯环稠合得到的芳环中的任意一种;具体地,r3为苯基,含杂原子取代基的苯基(如卤素取代的苯基)、对甲基苯基,对甲氧基苯基或杂芳基;
上述制备方法中,所述锰催化剂可为至少含有一个羰基的锰络合物。
所述锰催化剂具体可为:五羰基溴化锰和十羰基二锰中的至少一种。
所述格氏试剂可为:苯基溴化镁、对甲氧基苯基溴化镁和叔丁基氯化镁中的至少一种,其用料可为式ii所示脒的摩尔量的1-50%,具体可为5-25%或10%。
式iii所示炔烃与式ii所示脒的摩尔比可为1~5:1,具体可为2.5:1。
所述锰催化剂与式ii所示脒的摩尔比可为1:10~50,具体可为1:20。
所述c-h活化/c-c形成反应可在溶剂中进行,所述溶剂可为:1,4-二氧六环和/或四氢呋喃。
所述c-h活化/c-c形成反应的体系中,式ii所示脒的摩尔浓度可为0.05~1mol/l,具体可为0.25mol/l或0.5mol/l。
所述反应在惰性气氛下进行。
所述c-h活化/c-c形成反应的反应温度可为80~150℃,反应时间可为4~24h,具体可为在120℃的条件下反应12h。
本发明利用脒、炔烃于溶剂中在格氏试剂和锰催化剂的存在下进行反应,实现了多取代烯基脒的合成。多取代烯基脒是有机合成中重要的合成单元,现有的合成方法步骤繁琐,成本高昂。本发明使用一步法合成了多取代烯基脒骨架,底物适用性广,官能团容忍性好,在有机合成中具有广泛的应用前景。
附图说明
图1为本发明实施例1中制备(z)-n’-苯基-2-((e)-苯乙烯基)苯甲脒的反应方程式。
图2为本发明实施例2中制备(z)-4-甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒的反应方程式。
图3为本发明实施例3中制备(z)-4-三氟甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒的反应方程式。
图4为本发明实施例4中制备(z)-2-甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒的反应方程式。
图5为本发明实施例5中制备(z)-3,5-二甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒的反应方程式。
图6为本发明实施例6中制备(z)-2-((e)-4-甲氧基苯乙烯基)-n’-苯基苯甲脒的反应方程式。
图7为本发明实施例7中制备(z)-2-((e)-4-氟苯乙烯基)-n’-苯基苯甲脒的反应方程式。
图8为本发明实施例8中制备(z)-n’-苯基-2-((e)-2-(2-噻吩基)乙烯基)苯甲脒的反应方程式。
图9为本发明实施例9中制备(z)-n’-苯基-2-((e)-1-苯基-1-丙烯-2-烯基)苯甲脒的反应方程式。
具体实施方式
下面通过具体实施例对本发明进行说明,但本发明并不局限于此。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
实施例1、
根据图1所示的合成路线图制备(z)-n’-苯基-2-((e)-苯乙烯基)苯甲脒(式i-a)
在氮气保护下,向一个干燥的三口烧瓶内加入nah(60%分散于矿物油中)(15.0mmol,360mg),之后向反应瓶中加入5ml干燥的dmso,之后将反应体系置于冰浴中,并加以搅拌,随后将苯胺(11.0mmol,1.0g),苯甲腈(10mmol,1.0g)加入到反应体系中。在0℃下搅拌1h,之后在室温下搅拌12h,之后在剧烈搅拌下向反应体系中加入50ml冰水,淬灭后的反应体系使用乙酸乙酯(3*20ml)进行萃取,合并萃取液,并使用无水na2so4进行干燥,将干燥后的体系过滤,滤液旋干,经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:20:3),得到目标产物n-苯基苯甲脒(式ii-a)1.66g,产率85%。
1hnmr(400mhz,cdcl3):7.86(d,j=6.8hz,2h),7.47(m,3h),7.36(m,2h),7.07(t,j=7.2hz,1h),6.99(d,j=7.2hz,2h),4.84(br,2h)。
13cnmr(125hz,cdcl3):154.6,149.7,135.7,130.4,129.4,128.3,126.7,122.8,121.5,结构正确。
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),n-苯基苯甲脒(式ii-a)(1mmol,196mg),1,4-二氧六环(2ml),苯乙炔(式iii-a)(2.5mmol,255mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=100:10:3)后得到目标产物(式i-a)247mg,产率83%。
1hnmr(500mhz,cdcl3):7.73-7.57(m,3h),7.51(m,3h),7.36(m,4h),7.26m,2h),7.07(m,4h),4.85(s,2h)。
13cnmr(126mhz,cdcl3):155.7,149.5,137.4,136.0,135.6,130.7,129.7,128.8,128.6,128.0,127.9,127.6,126.7,126.4,126.2,123.2,121.7。
hrms(esi):calculatedforc21h19n2+([m+h]+):299.15428,found:299.15396,结构正确。
实施例2、
根据图2所示的反应方程式制备(z)-4-甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒(式i-b)
在氮气保护下,向一个干燥的三口烧瓶内加入nah(60%分散于矿物油中)(15.0mmol,360mg),之后向反应瓶中加入5ml干燥的dmso,之后将反应体系置于冰浴中,并加以搅拌,随后将苯胺(11.0mmol,1.0g),4-甲基苯甲腈(10mmol,1.2g)加入到反应体系中。在0℃下搅拌1h,之后在室温下搅拌12h,之后在剧烈搅拌下向反应体系中加入50ml冰水,淬灭后的反应体系使用乙酸乙酯(3*20ml)进行萃取,合并萃取液,并使用无水na2so4进行干燥,将干燥后的体系过滤,滤液旋干,经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:20:3),得到目标产物4-甲基-n-苯基苯甲脒(式ii-b)1.79g,产率85%。
1hnmr(400mhz,cdcl3)δ7.78(d,j=7.6hz,2h),7.35(m,2h),7.25(d,j=7.6hz,2h),7.06(t,j=7.6hz,1h),6.99(d,j=7.6hz,2h),4.79(br,2h),2.41(s,3h)。
13cnmr(125hz,cdcl3)δ154.5,149.9,140.6,132.9,129.4,129.1,126.6,122.8,121.6,21.3。结构正确。
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),4-甲基-n-苯基苯甲脒(式ii-b)(1mmol,210mg),1,4-二氧六环(2ml),苯乙炔(式iii-a)(2.5mmol,255mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-b)284mg,产率91%。
1hnmr(400mhz,cdcl3):7.64(d,j=14.9hz,1h),7.49(d,j=7.4hz,4h),7.35(t,j=7.5hz,4h),7.27-7.23(m,1h),7.10-7.02(m,5h),4.82(s,2h),2.39(s,3h)。
13cnmr(101mhz,cdcl3)139.5,137.5,135.4,130.4,129.6,128.8,128.6,128.4,127.8,126.7,123.0,121.7,21.5。
hrms(esi):calculatedforc22h21n2+([m+h]+):313.16993,found:313.16934,结构正确。
实施例3、
根据图3所示的合成路线图制备(z)-4-三氟甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒(式i-c)
在氮气保护下,向一个干燥的三口烧瓶内加入nah(60%分散于矿物油中)(15.0mmol,360mg),之后向反应瓶中加入5ml干燥的dmso,之后将反应体系置于冰浴中,并加以搅拌,随后将苯胺(11.0mmol,1.0g),4-三氟甲基苯甲腈(10mmol,1.7g)加入到反应体系中。在0℃下搅拌1h,之后在室温下搅拌12h,之后在剧烈搅拌下向反应体系中加入50ml冰水,淬灭后的反应体系使用乙酸乙酯(3*20ml)进行萃取,合并萃取液,并使用无水na2so4进行干燥,将干燥后的体系过滤,滤液旋干,经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:20:3),得到目标产物4-三氟甲基-n-苯基苯甲脒(式ii-c)1.26g,产率48%。
1hnmr(500mhz,cdcl3):4.89(brs,2h,2×nh),6.98(d,j=7.4hz,2h,ar),7.09(t,j=7.4hz,1h,ar),7.37(dd,j=7.4,7.4hz,2h,ar),7.70(d,j=8.0hz,2h,ar),7.99(d,j=8.0hz,2h,ar)。
13cnmr(125mhz,cdcl3):121.3(2c),122.8,123.8(q,j=272.3hz),125.5(q,j=3.6hz,2c),127.2(2c),129.6(2c),132.4(q,j=32.4hz),139.2,149.3,153.3。结构正确。
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),4-三氟甲基-n-苯基苯甲脒(式ii-c)(1mmol,264mg),1,4-二氧六环(2ml),苯乙炔(式iii-a)(2.5mmol,255mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-c)263mg,产率72%。
1hnmr(400mhz,cdcl3):7.94(s,1h),7.70(d,j=7.5hz,1h),7.66-7.54(m,1h),7.52(d,j=7.9hz,3h),7.40-7.36(m,5h),7.32-7.29(m,1h),7.11-7.08(m,1h),7.02-7.01(m,2h),4.92(s,2h)。
13cnmr(101mhz,cdcl3):154.6,148.9,138.9,136.8,136.5,132.4,129.8,129.3,128.9,128.5,128.4,126.9,124.9,124.0,123.5,123.0,121.4。
19fnmr(565mhz,cdcl3):-62.83。
hrms(esi):calculatedforc22h18f3n2+([m+h]+):367.14166,found:367.14093,结构正确。
实施例4、
根据图4所示的合成路线图制备(z)-2-甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒(式i-d)
在氮气保护下,向一个干燥的三口烧瓶内加入nah(60%分散于矿物油中)(15.0mmol,360mg),之后向反应瓶中加入5ml干燥的dmso,之后将反应体系置于冰浴中,并加以搅拌,随后将苯胺(11.0mmol,1.0g),2-甲基苯甲腈(10mmol,1.2g)加入到反应体系中。在0℃下搅拌1h,之后在室温下搅拌12h,之后在剧烈搅拌下向反应体系中加入50ml冰水,淬灭后的反应体系使用乙酸乙酯(3*20ml)进行萃取,合并萃取液,并使用无水na2so4进行干燥,将干燥后的体系过滤,滤液旋干,经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:20:3),得到目标产物2-甲基-n-苯基苯甲脒(式ii-d)1.55g,产率74%。
1hnmr(400mhz,cdcl3):7.50(d,j=7.6hz,1h),7.37(m,2h),7.31(d,j=7.6hz,1h),7.26(m,1h),7.08(m,4h),4.74(br,2h),2.57(s,3h)。
13cnmr(125hz,cdcl3):156.0,149.4,136.8,135.7,130.7,129.4,129.0,127.7,125.7,122.9,121.5,19.5。结构正确。
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),2-甲基-n-苯基苯甲脒(式ii-d)(1mmol,210mg),1,4-二氧六环(2ml),苯乙炔(式iii-a)(2.5mmol,255mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-d)209mg,产率67%。
1hnmr(500mhz,cdcl3):7.46(s,1h),7.38-7.34(m,3h),7.28-7.22(m,6h),7.04-6.99(m,5h),4.76(s,2h),2.54(s,3h)。
13cnmr(126mhz,cdcl3):156.2,149.5,137.0,135.8,130.8,129.6,129.2,128.8,127.8,126.6,125.8,123.0,121.6,19.7。
hrms(esi):calculatedforc22h21n2+([m+h]+):313.16993,found:313.16928,结构正确。
实施例5、
根据图5所示的合成路线图制备(z)-3,5-二甲基-n’-苯基-2-((e)-苯乙烯基)苯甲脒(式i-e)
在氮气保护下,向一个干燥的三口烧瓶内加入nah(60%分散于矿物油中)(15.0mmol,360mg),之后向反应瓶中加入5ml干燥的dmso,之后将反应体系置于冰浴中,并加以搅拌,随后将苯胺(11.0mmol,1.0g),3,5-二甲基苯甲腈(10mmol,1.3g)加入到反应体系中。在0℃下搅拌1h,之后在室温下搅拌12h,之后在剧烈搅拌下向反应体系中加入50ml冰水,淬灭后的反应体系使用乙酸乙酯(3*20ml)进行萃取,合并萃取液,并使用无水na2so4进行干燥,将干燥后的体系过滤,滤液旋干,经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:20:3),得到目标产物3,5-二甲基-n-苯基苯甲脒(式ii-e)1.68g,产率75%。
1hnmr(500mhz,cdcl3):δ7.48(s,2h),7.34(t,j=7.6hz,2h),7.10(s,1h),7.05(t,j=7.3hz,1h),6.98(d,j=7.5hz,2h),4.80(s,2h),2.36(s,6h)。
13cnmr(126mhz,cdcl3):δ154.9,150.0,138.2,135.8,132.2,129.5,124.5,122.90,121.6,21.3,结构正确。
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),3,5-二甲基-n-苯基苯甲脒(式ii-e)(1mmol,224mg),1,4-二氧六环(2ml),苯乙炔(式iii-a)(2.5mmol,255mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-e)104mg,产率32%。
1hnmr(400mhz,cdcl3):7.49-7.47(m,2h),7.39-7.19(m,7h),7.06-6.94(m,5h),4.74(s,2h),2.36-2.33(m,6h)。
13cnmr(101mhz,cdcl3):137.5,136.9,136.9,133.7,132.2,132.1,129.6,129.5,129.1,128.7,128.0,127.7,127.4,126.4,125.4,122.9,121.6,21.0,20.8。
hrms(esi):calculatedforc23h23n2+([m+h]+):327.18558,found:327.18500,结构正确。
实施例6、
根据图6所示的合成路线图制备(z)-2-((e)-4-甲氧基苯乙烯基)-n’-苯基苯甲脒(式i-f)
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),n-苯基苯甲脒(式ii-a)(1mmol,196mg),1,4-二氧六环(2ml),4-甲氧基苯乙炔(式iii-b)(2.5mmol,330mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:20:3)后得到目标产物(式i-f)272mg,产率83%。
1hnmr(500mhz,cdcl3):7.67(d,j=7.2hz,1h),7.59(d,j=5.7hz,1h),7.53-7.34(m,6h),7.32-7.25(m,1h),7.08-7.02(m,3h),6.90-6.88(m,3h),4.85(s,2h),3.81(s,3h)。
13cnmr(126mhz,cdcl3):159.5,155.9,149.5,135.9,135.8,130.3,129.7,129.1,128.9,128.6,128.0,127.9,127.3,125.9,124.2,123.1,121.7,55.4。
hrms(esi):calculatedforc22h21on2+([m+h]+):329.16484,found:329.16425,结构正确。
实施例7、
根据图7所示的合成路线图制备(z)-2-((e)-4-氟苯乙烯基)-n’-苯基苯甲脒(式i-g)
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),n-苯基苯甲脒(式ii-a)(1mmol,196mg),1,4-二氧六环(2ml),4-氟苯乙炔(式iii-c)(2.5mmol,300mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-g)271mg,产率86%。
1hnmr(500mhz,cdcl3):7.71-7.54(m,3h),7.49-7.28(m,6h),7.08-7.02(m,6h),4.87(s,2h)。
13cnmr(126mhz,cdcl3):163.5,161.5,155.8,149.3,135.6(d,1jc-f=236),133.6,131.0,129.7,129.5,128.6,128.2(d,2jc-f=30),127.6,126.1,123.3,121.6,115.8,15.6。
19fnmr(565mhz,cdcl3):-113.87。
hrms(esi):calculatedforc21h18fn2+([m+h]+):317.14485,found:317.14433,结构正确。
实施例8、
根据图8所示的合成路线图制备(z)-n’-苯基-2-((e)-2-(2-噻吩基)乙烯基)苯甲脒(式i-h)
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),n-苯基苯甲脒(式ii-a)(1mmol,196mg),1,4-二氧六环(2ml),2-噻吩基乙炔(式iii-d)(2.5mmol,270mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-h)213mg,产率70%。
1hnmr(500mhz,cdcl3):7.60(dj=41.3hz,2h),7.49(d,j=16.1hz,1h),7.41-7.21(m,8h),7.07-7.02(m,3h),4.86(s,2h)。
13cnmr(126mhz,cdcl3):155.8,149.5,140.3,135.8,135.6,129.7,129.6,128.6,127.5,126.4,126.3,125.9,125.0,124.8,123.2,122.8,121.6。
hrms(esi):calculatedforc19h17sn2+([m+h]+):305.11070,found:305.11017,结构正确。
实施例9、
根据图9所示的合成路线图制备(z)-n’-苯基-2-((e)-1-苯基-1-丙烯-2-烯基)苯甲脒(式i-i)
在氮气保护下,向干燥的50mlschlenk瓶中依次加入催化剂五羰基溴化锰(0.05mmol,14mg),n-苯基苯甲脒(式ii-a)(1mmol,196mg),1,4-二氧六环(2ml),1-苯基丙炔(式iii-e)(2.5mmol,290mg)和苯基溴化镁(0.1ml)(1mol/l的四氢呋喃溶液),并在120℃的温度下反应12h。反应后向反应体系中加入10ml饱和氯化铵水溶液将反应体系进行淬灭,之后使用乙酸乙酯(3×10ml)萃取,合并有机相,使用无水硫酸镁进行干燥,将干燥后的体系过滤,滤液在真空条件下旋干。经柱色谱提纯(洗脱剂为石油醚:乙酸乙酯:三乙胺=80:10:3)后得到目标产物(式i-i)165mg,产率52%。
1hnmr(500mhz,cdcl3):7.74(bs,1h),7.39-7.30(m,9h),7.25-7.21(m,1h),7.01-6.94(m,3h),6.61(bs,1h),4.89(s,2h),2.32(s,3h)。
13cnmr(126mhz,cdcl3):156.4,149.5,143.8,139.1,137.6,135.0,129.6,129.4,129.3,129.1,128.9,128.8,128.2,127.3,126.6,122.8,121.4,19.8。
hrms(esi):calculatedforc22h21n2+([m+h]+):313.16993,found:313.16943,结构正确。
- 还没有人留言评论。精彩留言会获得点赞!