FIPV基因过量表达的转基因拟南芥株系及其构建方法与流程
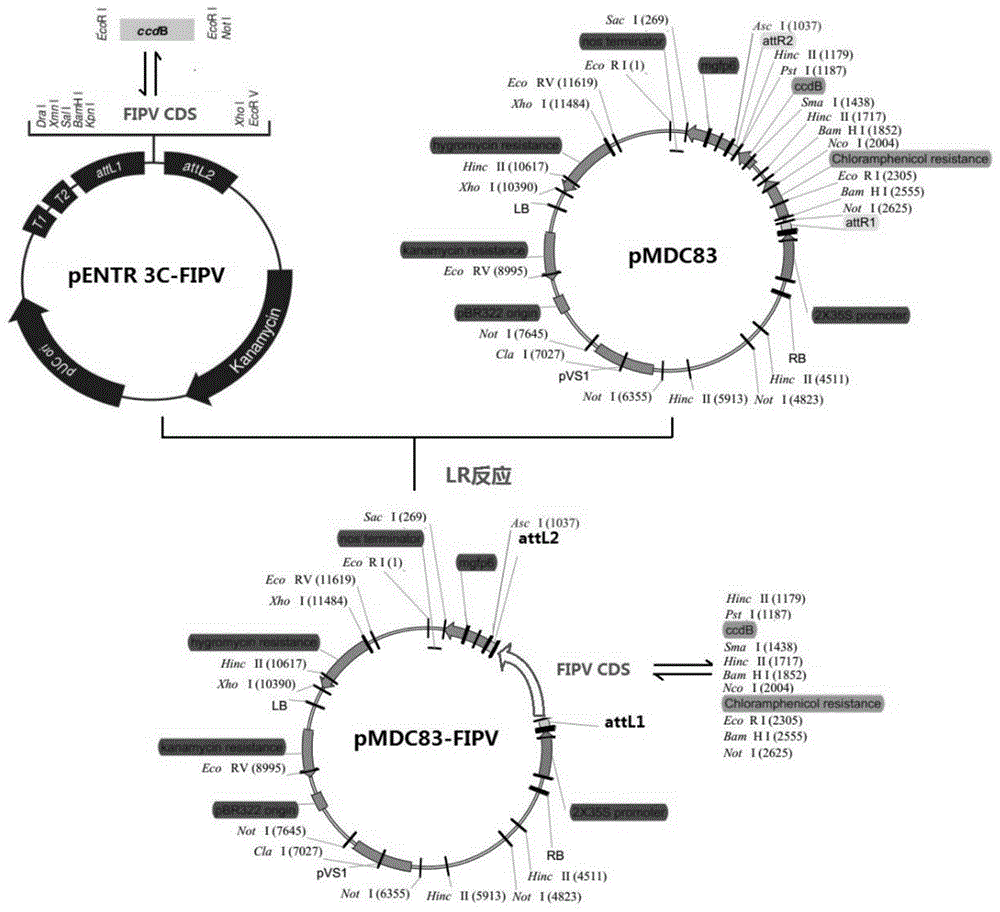
本发明属于植物转基因
技术领域:
,具体涉及一种fipv基因过量表达的转基因拟南芥株系及其构建方法。
背景技术:
:氮素对植物的生长和发育起着非常重要的作用,绝大多数的陆生植物吸收氮素以硝态氮(no3-)为主。大量的研究表明,no3-不但是一种营养物质,而且还是一种信号分子,在植物短期和长期生长过程中起调控作用。短期效应主要体现在硝态氮初级响应,即施加no3-时植物体内有超过1000多个基因的表达迅速发生变化,比如nrts,nias和nir在no3-处理几分钟内被诱导表达。长期效应表现在no3-影响植物根系形态建成、种子休眠、开花、生物钟和不依赖于aba的气孔关闭和生长素运输等过程。近十年来,一些重要的参与no3-初级响应过程并影响no3-代谢过程的调控基因被鉴定。这些调控基因包括转运蛋白类,如nrt1.1;蛋白激酶类,如cipk8和cipk23;转录因子类,如nlp7、tga1、tga4、tcp20、nrg2等;microrna类,如micr167。最近的研究表明,参与真核生物mrna前体3’末端加工的多聚腺苷酸化复合体(cpsf)在调控硝态氮信号途径中起重要作用。fipv是cpsf的关键成员,极有可能调控植物的硝态氮感知、吸收、转运和同化等过程。因此鉴定该基因的功能对于研究植物硝态氮代谢过程,进而提高植物氮素利用率非常重要。鉴定基因功能首先需要获得该基因的突变体和过量表达株系。在突变体中由于缺失该基因,会产生相应的分子、生化或者生理表型,由此可以推断出该基因的功能。同时,还需要研究该基因大量表达时,分子、生化或者生理表型的变化。因此,构建目的基因的过量表达株系是鉴定基因功能的必需环节。一般地,利用拟南芥构建目的基因过量表达株系采用根瘤农杆菌介导的浸花法。首先,提取植物的总rna,以此为模板进行反转录得到第一链cdna,进行pcr反应克隆目的基因的cdna或者编码区序列(cds)。然后,将克隆得到的cdna或cds连入强启动子(如35spromoter)下游构建植物表达载体,将该表达载体转化根瘤农杆菌。最后,将转化的农杆菌培养至合适浓度,将拟南芥的花序侵入其中,农杆菌可以通过花粉管进入植物受体,并将包含强启动子融合目的基因cdna或cds的序列整合到宿主植物基因组中。植物表达载体一般都含有抗生素标记基因(如抗kana,氨苄或潮霉素基因)用以筛选转基因植物。转基因植株的种子可以生长在含有相应抗生素的固体培养基上并萌发和生长,非转基因种子只能萌发而不能生长。研究者一般希望过量表达株系中的目的基因表达量尽量高并且能够稳定遗传。因此,研究fipv基因的功能,构建高效表达且稳定遗传的过量表达系是鉴定该基因在调控no3-代谢途径中的功能是关键关节。技术实现要素:为了达到目的基因高效表达和稳定遗传的目的,本发明提供了一种fipv基因过量表达的转基因拟南芥株系及其构建方法。为了实现上述目的,本发明采用的技术方案为:本发明fipv基因过量表达的转基因拟南芥株系的构建方法,所述fipv基因过量表达的转基因拟南芥株系的构建方法包括:用根瘤农杆菌通过浸花法得fipv基因过量表达的转基因拟南芥株系。所述fipv基因过量表达的转基因拟南芥株系的构建方法包括以下步骤:步骤一,拟南芥培养,将4℃低温和避光处理3d的拟南芥种子播种于蛭石上,加入1/2ms培养液,置于相对湿度70%,温度23℃,光照和黑暗时间分别为16h和/8h的植物专用培养箱中培养;步骤二,制备大肠杆菌dh5α感受态细胞;步骤三,利用gateway技术构建fipv过表达的表达载体pmdc83-fipv;步骤四,利用试剂盒法提取pmdc83-fipv质粒;步骤五,制备农杆菌gv3101感受态细胞;步骤七,pmdc83-fipv表达载体转化农杆菌;步骤八,筛菌鉴定阳性农杆菌转化子;步骤九,农杆菌介导的遗传转化;步骤十,转基因植株t1代的获得及分离比的鉴定;步骤十一,转基因植株t3代中筛选纯合的fipv过表达转基因株系;步骤十一,fipv过表达转基因株系的dna水平、rna水平及蛋白水平的鉴定。所述步骤三具体包括:提取拟南芥总rna,然后进行反转录得到第一链cdna,以此为模板,设计特异扩增fipv编码区序列(cds)的正向引物f和反向引物r,两引物的序列如下:f(正向引物):5’-cggggtaccatggaagaggacgatgag-3’;r(反向引物):5’-ccgctcgagtcatcccacacatactct-3’;利用f和r进行pcr反应,产物凝胶回收后通过酶切连接入gateway兼容的载体pentr3c,构建克隆载体pentr3c-fipv,然后将pentr3c-fipv与含有35s强启动子和潮霉素标记基因的表达载体pmdc83进行lr反应,得到fipv过表达的pmdc83-fipv表达载体。用该表达载体转化感受态大肠杆菌dh5α,对pmdc83-fipv序列测定后得到拟南芥fipv基因cds序列所示:用限制性内切酶kpni和xhoi分别对pcr产物和gateway兼容的载体pentr3c进行双酶切,将得到的含有黏性末端的fipvcds连接入酶切开的pentr3c构建克隆载体pentr3c-fipv,然后将pentr3c-fipv与包含35s强启动子和潮霉素标记基因的表达载体pmdc83进行lr反应,得到fipv过表达的pmdc83-fipv表达载体。首次克隆到了拟南芥fipv基因的编码区序列,具体序列如下:本发明所述fipv基因过量表达的转基因拟南芥株系为oe31和oe51。本发明的有益效果为:1.本发明构建的fipv基因过表达拟南芥转基因株系为首次报道,可以直接用农杆菌介导的浸花法进行遗传转化,获得fipv过表达的新种质,为该鉴定该基因的功能鉴定提供基础。2.构建过量表达系所使用的植物表达载体在目的基因下游融合一个gfp标记基因。因此,该过量表达株系除了实现目的基因高效表达外,gfp蛋白还可以用于fipv基因在植物中的亚细胞定位。同时,使用gfp抗体通过免疫反应可以提取过表达株系中fipv融合gfp蛋白,实现提纯fipv蛋白的目的。3.在筛选阳性的转化农杆菌克隆的中,使用抗生素kana,而在筛选阳性转基因植株中,使用抗生素潮霉素。使用两种不同的抗生素最为筛选条件,可以极大减少假阳性出现的几率,确保得到的阳性克隆特异性高。附图说明图1为植物表达载体pmdc83-fipv构建方法示意图;图2为以拟南芥cdna为模板克隆fipv基因cds的电泳图,其中,1—4泳道3792bp处条带为fipv基因cds;图3为以大肠杆菌中克隆载体pentr3c-fipv(重组质粒)为模板的克隆fipv基因cds的电泳图,其中,1—4泳道3792bp处条带为fipv基因cds,5泳道为不加模板的阴性对照;图4为以大肠杆菌中表达载体pmdc83-fipv(重组质粒)为模板的克隆fipv基因cds的电泳图,其中,1—4泳道3792bp处条带为fipv基因cds,5泳道为不加模板的阴性对照;图5为以根瘤农杆菌中表达载体pmdc83-fipv(重组质粒)为模板的克隆fipv基因cds的电泳图,其中,1—4泳道3792bp处条带为fipv基因cds,5泳道为不加模板的阴性对照;图6为利用荧光实时定量pcr检测fipv过表达的转基因株系oe31和oe51中fipv表达量。具体实施方式下面结合实例对本发明做进一步说明。实施例1拟南芥fipv基因cds序列的克隆,植物材料为拟南芥。1.拟南芥总rna的提取(试剂盒法)利用试剂盒法(柱式总rna提取试剂盒)提取相应拟南芥植株的总rna,具体方法如下:(1)称取约100mg材料于1.5mleppendorf管中,在液氮中充分研磨;(2)加入1mlbufferrlt(材料体积小于等于bufferrlt体积的10%),充分震荡混匀,室温静置5min;(3)为充分去除细胞壁残渣、蛋白、脂肪、多糖等,4℃,12000rpm离心10min,将上清转移到新的离心管中;(4)分相:①加0.2ml氯仿,剧烈震荡15s,室温静置2min;②离心(4℃,12000rpm,勿超过12000rpm)10min;(5)沉淀,并去除多糖:①取无色水相(大约原初bufferrlt体积的50%,约500μl)至一新的eppendorf管中,切勿吸到中间层;②加0.25ml70%乙醇,颠倒混匀;(6)将上步所得溶液加入到已装入吸附柱的收集管中(若液体体积超过700μl,则分两次转移),4℃,12000rpm离心20s,倒掉收集管中的废液,将吸附柱重新放回收集管中;(7)向吸附柱中加入700μlbufferrw1,4℃,12000rpm离心20s,倒掉收集管中的废液,将吸附柱重新放回收集管中;(8)向吸附柱中加入500μlbufferrw2,4℃,12000rpm离心20s,倒掉收集管中的废液,将吸附柱重新放回收集管中;(9)重复步骤(8)(10)4℃,12000rpm空离2min;(11)将空的吸附柱置于室温数分钟,以彻底晾干;(12)将吸附柱置于一个新的无rnase离心管中,加入30μlrnase-freewater,室温放置1min,4℃,12000rpm离心1min,收集rna溶液,置于-70℃保存。2.反转录取1μg总rna,加oligd(t)0.5μl,补充rnase-freewater至总体积为6μl。65℃水浴中放置5min,稍离心后在管中配置下列反转录反应液。42℃保温1h后,70℃处理15min,冰上冷却即可。3.利用phusion高保真dna聚合酶进行pcr扩增(1)取0.2mlpcr专用管,依次加入以下成分:(2)封闭pcr管,放入pcr仪中。反应条件为:98℃预变性2min,98℃变性20s,根据引物的退火温度退火20s,72℃延伸30s/kb,循环30次,72℃后延伸10min。(3)反应完毕后,在1%琼脂糖凝胶中电泳,检查扩增效果。pcr产物琼脂糖凝胶电泳检测结果如图2所示,在3792bp处可以观察到清晰的目的条带。实施例2克隆载体pentr3c-fipv的构建1.pcr扩增fipv基因cds并与pentr3c连接f(正向引物):5’-cggggtaccatggaagaggacgatgag-3’;r(反向引物):5’-ccgctcgagtcatcccacacatactct-3’;以提取的cdna为模板,通过特异性引物f和r进行pcr扩增,并在目的基因上游和下游分别引入kpni和xhoi酶切位点。用天根(北京)胶回收试剂盒回收pcr产物,pcr产物与gateway兼容的pentr3c进行酶切连接,操作步骤按下表进行。成分用量目的片段回收产物0.5-10ngpent3c0.5μlsaltsolution0.5μlh2oupto3μl混匀后,室温(25℃)下反应30分钟,然后冰浴1分钟,用于转化大肠杆菌dh5α,表面涂含有卡那霉素的lb培养平板,37℃培养12小时。然后,挑取单个白色克隆涂布新的含有卡那霉素的lb培养平板,37℃培养12小时,进行菌落pcr扩增,如图3所示。同时,将筛菌正确的菌株送上海生工公司进行dna测序。dnaman软件将测序序列与拟南芥fipv基因启动子序列进行比对,比对正确后说明克隆载体pentr3c-fipv构建成功,提取阳性质粒备用。质粒的提取使用北京康维公司试剂盒,按说明书步骤进行。(1)取1-5ml过夜培养的菌液,加入离心管中,12000rpm离心1min,尽量吸弃上清。(2)向留有菌体沉淀的离心管中加入250μlbufferp1,使用移液器或涡旋振荡器彻底悬浮细菌沉淀。注意:如果菌块未彻底混匀,将会影响裂解效果,使提取量和纯度偏低。(3)向离心管中加入250μlbufferp2,温和地上下颠倒混匀4-6次,使菌体充分裂解,此时菌液应变得清亮粘稠。所用时间不应超过5min,以免质粒受到破坏。(4)向离心管中加入350μlbuffern3,立即温和地上下颠倒混匀4-6次,此时将出现白色絮状沉淀,12000rpm离心10min,此时在离心管底部形成沉淀。(5)柱平衡:向已装入吸附柱(spincolumn)的收集管中加入200μlbufferps,12000rpm离心2min,倒掉收集管中的废液,将吸附柱重新放回收集管中。(6)将步骤4中所得上清加入到已装入吸附柱的收集管中,注意不要吸出沉淀,12000rpm离心1min,倒掉收集管中的废液,将吸附柱放回收集管中。(7)向收集管中加入600μlbufferpw,12000rpm离心1min,倒掉收集管中的废液。(8)重复步骤7。(9)将吸附柱重新放回收集管中,12000rpm离心1min,倒掉收集管中的废液。将吸附柱置于室温数分钟,以彻底晾干。(10)将吸附柱置于一个新的离心管中,向吸附膜的中间部位悬空滴加50-100μlbuffereb,室温放置数分钟,12000rpm离心1min,将质粒溶液收集到离心管中。-20℃保存质粒。实施例3植物表达载体pmdc83-fipv的构建将测序正确的克隆载体pentr3c-fipv与含有35s启动子和gfp报告基因的pmdc83进行lr进行反应,得到35s启动子驱动的fipv基因融合gfp报告基因的植物表达载体。lr反应体系如下:克隆载体1μl(约100ng)载体,pmdc83载体1μl(约100ng),lr克隆酶0.5μl。将上述物质混匀后置于室温(25℃)反应1-2小时,转化大肠杆菌dh5α。转化的菌株表面涂含有卡那霉素的lb培养平板,37℃培养12小时。然后,挑取单个白色克隆涂布新的含有卡那霉素的lb培养平板,37℃培养12小时,进行菌落pcr扩增,如图4所示。说明表达载体pmdc83-fipv构建成功,提取阳性质粒备用,提取方法如实施例2所述。实施例4fipv过表达的拟南芥转基因株系构建1.根癌农杆菌gv3101感受态细胞的制备与转化农杆菌感受态细胞的制备方法如下所示:(1)挑取单菌落gv3101,接种于5mlyep液体培养基中,28℃,200rpm过夜培养。(2)取2ml培养物至50ml液体yep中,继续培养至od600为0.5左右。(3)将培养物冰浴30min,4℃,5000rpm,离心5min。(4)弃去上清,10ml0.1mol/l冷的nacl悬浮菌体;4℃,5000rpm,离心5min。(5)弃上清,沉淀用1ml20mmol/l冷的cacl2悬浮,分装成200μl/管,液氮中冷冻后-70℃保存。表达载体转化农杆菌采用冻融法,转化过程如下:(1)从-80℃取出gv3101感受态细胞(250μl),冰上解冻,立即加入1-2μl质粒dna。(2)冰上静置5min。(3)液氮中冷冻5min。(4)37℃水浴热激5min。(5)加入无抗生素的yep液体培养基1ml,28℃摇床培养2-4h。(6)4000rpm室温离心5min,收集菌体,用80-100μlyep溶液重新悬浮菌体。(7)将菌液均匀涂布于yep固体选择培养基,28℃静置培养2-3天。(8)长出菌落后,挑取单菌落重新划线于含抗生素的yep固体平板上,36h后进行菌落pcr鉴定,结果如图5所示。2.拟南芥的转化拟南芥的遗传转化采用花序浸染法,方法如下:(1)将拟南芥的主花序的剪掉,以诱导侧花序的生成,且侧花序同时开花,便于转化。转化前将材料浇透营养液,转化后要控制浇水或营养液,以便于种子及时成熟。(2)提前一天准备好转化所需要用的农杆菌,取2-5ml过夜培养的菌液加到250ml的培养基中摇床培养,以便于第二天可用于转化拟南芥。(3)收集过夜培养的农杆菌菌体,重新悬浮于50-100ml浸染液中。(4)将拟南芥伸长的种荚和已经开放的花全部剪掉后,将花絮浸入浸染液中,上下晃动花絮,以利于浸染液的进入。浸染时间一般持续1h左右。将浸染后的拟南芥平放入大盆中,薄膜覆盖以保湿,同时外罩黑色薄膜避光。黑暗处理后后将拟南芥取出置于光下,待成熟后收种子。拟南芥浸染液配方如下:用1mkoh调ph值至5.7。3.转基因株系的筛选收集侵染后的拟南芥t0代种子,进行消毒处理,方法如下:(1)取适量种子于1.5mleppendorf管中,标明相关信息;(2)事先配置好70%乙醇,用移液器取1ml于管中,吸打混匀,使种子可以充分地接触乙醇溶液,静置处理5min(注意将管壁和管盖上的种子都冲洗下来以保证灭菌彻底),之后用1ml移液器将离心管内的乙醇吸出(注意不要将种子吸出);(3)吐温水冲洗一遍;(4)用移液器取1ml2.6%次氯酸钠溶液于管中,上下颠倒混匀,静置处理10min;(5)吐温水冲洗五遍;(6)置于4℃保存待用。转基因植株的筛选将消毒后的t0代种子点播在含有50μg/ml潮霉素的ms固体培养基上,在植物专用培养室中生长7d,培养条件为22℃,长日照(16h光照/8h黑暗)。在t1代中挑选能够在潮霉素ms平板上萌发和生长的植株,分别转移到蛭石上培养,培养条件同上。收集t1代种子,按上述方法消毒,点播在含有50μg/ml潮霉素的ms固体培养基上,在植物专用培养室中生长7d,培养条件为22℃,长日照(16h光照/8h黑暗)。在t2代中挑选能够在潮霉素ms平板上萌发和生长的植株,分别转移到蛭石上培养,培养条件同上。收集t2代种子,按上述方法消毒,点播在含有50μg/ml潮霉素的ms固体培养基上,在植物专用培养室中生长7d,培养条件为22℃,长日照(16h光照/8h黑暗)。在t3代中挑选能够在潮霉素ms平板上全部萌发和生长且不出现分离的植株,得到过量表达系oe31和oe51。实施例5fipv过表达的拟南芥转基因株系中fipv表达量检测利用实时荧光定量pcr(qrt-pcr)检测fipv过表达的转基因拟南芥株系oe31和oe51中fipv的表达量。将在oe31和oe51中提取的rna进行反转录后得到的cdna样品稀释至0.5-2ng/μl,反应体系如下表所示:封闭pcr管,放入荧光定量pcr仪中(abi7500fast)。反应条件为:先50℃预变性20s,再95℃预变性10s,再95℃变性15s,60℃退火1min,之后进行plateread,从步骤3开始循环40次,溶解曲线为95℃15s,60℃1min,95℃30s,60℃15s。反应以tublin2作为内参基因以保证结果的准确性,每个处理有三次生物学重复,三个系统重复。fipv过量表达系oe31和oe51中fipv的表达量如图6所示,结果表明oe31和oe51中fipv的表达量显著高于野生型和突变体,fipv在过量表达系中可以高效表达。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1