一种纳米UiO-67-NH2及其制备方法和应用与流程
本发明涉及纳米生物医药材料技术领域,具体涉及一种纳米uio-67-nh2及其制备方法和应用。
背景技术:
金属有机骨架(mofs)是一种晶态多孔物质,由金属离子和有机配体自组装形成三维(3d)网络结构。与传统的载药材料相比,mof具有比表面积大,孔隙率高,易于化学剪裁和改性,结构丰富多样,载药率高等传统的载药材料无法比拟的优点。
金属有机骨架用作喜树碱载体的例子很少,文献中有记录,把喜树碱负载到zif-8上,但由于zif-8孔径小,该载体的载药率仅为2%。拓扑替康是喜树碱的衍生物,mil-100负载拓扑替康的载药率为33%。
鉴于每年有数百万人被诊断出患有癌症,癌症治疗的方法变得越来越重要。化疗是一种常规的癌症治疗方法。喜树碱(cpt)对于多种恶性肿瘤,例如结肠癌,卵巢癌,肝癌和骨癌等都有很强的抗肿瘤活性。不幸的是,这种药物水溶性差,毒性副作用严重,生理条件下内酯环快速水解。纳米给药系统已被用于克服上述这些缺点。
在过去的二十年中,已经合成了各种类型的纳米给药系统,如无机介孔二氧化硅,聚合物胶束和脂质体等。然而,这些传统的纳米给药系统由于存在低载药率和不受控制的药物突释等缺点,其应用受到限制。申请号为201210552129.4的中国专利,公开了氧化石墨烯的表面增强拉曼散射定位药物载体及其制备方法,其采用氧化石墨烯与金属纳米粒子的复合结构装载药物,但是,由于石墨烯成本较高,且工艺复杂难以大规模实现。申请号为200710015042.2的中国专利,公开了喜树碱-层状双金属氢氧化物纳米复合物的制备方法,但是,由于其载药率最高情况下仅能达到20%,难以满足现阶段的需求。申请号为201610311302.x的中国专利,公开了一种纳米级金属有机骨架材料的制备方法与应用,其粒径为400-500nm,用于装载姜黄素,反应5天的载药率为93.1%,但其粒径过大,对于肿瘤的穿透性差,不适合用于装载抗癌药物,难以满足现阶段抗癌药物的装载及释放需求。
uio-67是一种重要的金属有机骨架,其毒性低,具有较好的热稳定性,还具有较大的孔径和较高的比表面,可以装载一些较大的药物分子,因此,uio-67引起了研究人员的关注。但是,uio-67存在的问题是,其水稳定性较差,在水中容易坍塌,如果应用于载药很容易造成药物突释;因此,通过氨基官能团修饰使uio-67水稳定性提高,并通过纳米技术将uio-67-nh2纳米化,并用于负载喜树碱,对于提高水溶性差喜树碱的生物利用度,带来缓控释有着重要的意义。
技术实现要素:
基于上述存在问题,本发明的目的在于,提供一种纳米uio-67-nh2及其制备方法和应用,通过氨基官能团修饰使uio-67水稳定性提高,并通过纳米技术将uio-67-nh2纳米化,再将其应用于负载喜树碱,制成抗癌药物,克服了传统给药系统的低载药率和不受控制的药物突释等缺点。
为此本发明提供了如下的技术方案:
一种纳米uio-67-nh2是以锆盐和有机羧酸为原料采用溶剂热法制备,并通过使用调节剂调节其晶体形貌而得到的纳米材料,其晶体结构如图1所示,形貌结构如图3所示;所述调节剂为冰醋酸、三乙胺、乙二胺任一种或它们的组合物。
进一步地,所述调节剂为冰醋酸、三乙胺按照13-16:1的体积比制成的组合物。所述调节剂组合使用的体积比在这个条件下,调节剂和溶剂的配比使得产品的粒度更为均一,且此时的产品加载药物时更为稳定。
进一步地,所述纳米uio-67-nh2的粒径范围在90~300nm之间,比表面积为240.14~300m2·g-1,孔容为415~450cm3·g-1。
本方案提供所述纳米uio-67-nh2的制备方法,采用溶剂热法,包括如下步骤:
(1)将锆盐和有机羧酸分别加入到溶剂中进行溶解,结合超声分散处理,分散后将两者在室温下混合搅拌15~25分钟,得到初混料,再向初混料中加入调节剂,得到混合原料;
(2)将所述混合原料加入到反应釜中,在118~125℃温度下反应22~26小时,得到初产物;
(3)将所述初产物离心分离后,采用真空干燥方式,于温度为100~105℃条件下干燥23~25小时,即得到纳米uio-67-nh2成品;
其中,所述锆盐和有机羧酸的摩尔比是75~79:95~102;所述溶剂和调节剂的重量比是145~155:12~16。
进一步地,所述锆盐和有机羧酸的摩尔比是77:100;所述溶剂和调节剂的重量比是150:15。
进一步地,所述锆盐是四氯化锆;所述有机羧酸是2-氨基-4,4’联苯二甲酸或2,2'-二吡啶-5,5'-二羧酸;所述溶剂为n,n-二甲基甲酰胺或n,n-二乙基甲酰胺;所述调节剂为冰醋酸、三乙胺、乙二胺任一种或它们的组合物。
进一步地,所述调节剂为冰醋酸、三乙胺按照13-16:1的体积比制成的组合物。所述调节剂组合使用的体积比在这个条件下,调节剂和溶剂的配比使得产品的粒度更为均一,且此时的产品加载药物时更为稳定。
进一步地,在步骤(2)中,所述反应温度为120℃,反应时间为24小时;所述反应釜为有聚四氟乙烯内衬的反应釜。在步骤(3)中,所述干燥温度为100℃,干燥时间为24小时。
本方案提供所述纳米uio-67-nh2的应用,是将uio-67-nh2用于装载药物,所述药物为喜树碱,且其载药率为36.53~38.23w%。
进一步地,具体是将uio-67-nh2与药物加入到甲醇溶剂中,于温度为22~28℃条件下搅拌装载5~7天;所述装载过程质量比为uio-67-nh2:药物=1:1~3。在这个条件下,uio-67-nh2与药物的装载率、载药效率最高。
进一步地,所述装载温度为25℃条件下,搅拌装载时间为7天。在这个条件下,uio-67-nh2与药物的装载率最高,且更为稳定。
其中,所有的原料例如:四氯化锆、2-氨基-4,4’联苯二甲酸、2,2'-二吡啶-5,5'-二羧酸、n,n-二甲基甲酰胺、n,n-二乙基甲酰胺、冰醋酸、三乙胺、乙二胺等都是从国内外的化学试剂公司进行购买,并没有经过继续提纯而是直接使用的。
uio-67是用4,4'-联苯二甲酸与zr4+组装的3d多孔材料,uio-67中的次级结构单元zr6o4(oh)4与12个有机配体配位形成3d结构,包括一个八面体中心孔笼和八个四面体角笼,因此,uio-67具有大孔径和高比表面积;但其水稳定性差,导致材料骨架容易塌陷,如果应用于载药则易造成药物突释。因此本方案发明人采用了溶剂热法,以锆盐和有机羧酸为原料通过配位作用,并通过使用调节剂调节其晶体形貌而得到的纳米材料。用于加载喜树碱,其载药率可达36.53~38.23w%,且可控制缓释放克服了传统给药系统的低载药率和不受控制的药物突释等问题。
本发明具有以下有益效果:
(1)本发明通过氨基官能团修饰使uio-67水稳定性提高,通过对uio-67配体修饰亲水性氨基(标记为uio-67-nh2),提高了其水稳定性;
(2)本发明通过改变添加剂的添加量,调节uio-67-nh2的粒径;本发明合成的uio-67-nh2纳米粒子平均粒径150nm,粒径分布图表明大多数粒子的尺寸小于200nm,且其粒径呈正态分布。sem图表明纳米颗粒是团聚的,这与pdi(pdi=0.80)结果描述的分散水平一致。纳米尺寸的uio-67-nh2载药体系将有利于增加药物溶出速度,保护不稳定药物,促进药物生物利用度,同时,100-200nm的粒子也易于渗透到肿瘤组织血管内皮细胞间隙。
(3)本发明纳米uio-67-nh2的比表面积为240.14~300m2·g-1,孔容为415~450cm3·g-1。较大的孔容积有助于装载尺寸较大的药物分子。
(4)本发明制备的纳米uio-67-nh2对喜树碱的载药率高达38.23w%,并具有持续、可控、缓释放的性能。
附图说明
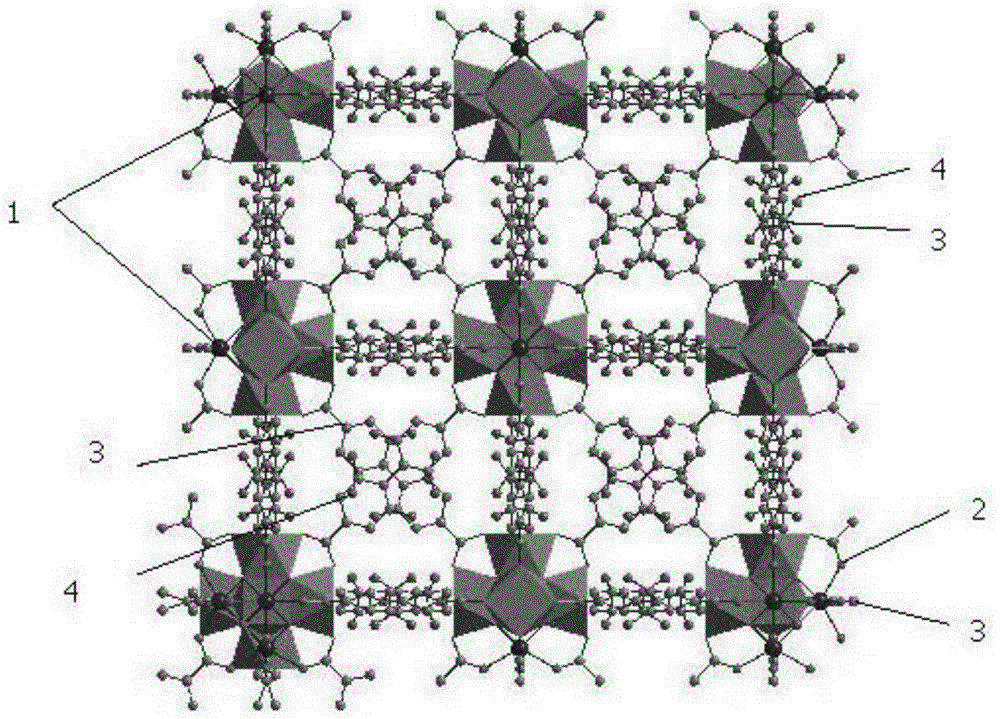
图1为实施例1中制备的纳米uio-67-nh2的晶体结构图;其中,1为zr,2为o,3为c,4为nh2。
图2为实施例1中制备的纳米uio-67-nh2的xrd图。
图3为实施例1中制备的纳米uio-67-nh2的sem图。
图4为实施例1中制备的纳米uio-67-nh2的n2吸附脱附等温线.
图5为实施例1中制备的纳米uio-67-nh2的孔径分布曲线。
图6为实施例1中制备的纳米uio-67-nh2的粒径分布图。
图7为实施例6中制备的纳米uio-67-nh2加载药物前的zeta电位测量图。
图8为实施例6中制备的纳米uio-67-nh2加载药物后的zeta电位测量图。
图9为实施例6中制备的纳米uio-67-nh2和加载药物后uio-67-nh2以及配体和cpt稳定性的ft-ir图。
图10为实施例6中制备的纳米uio-67-nh2稳定性的xrd图。
图11为实施例6中制备的纳米uio-67-nh2加载药物cpt后在磷酸盐缓冲液的药物释放曲线图。
具体实施方式
下面通过实施例对本发明作进一步说明,但不作为是对本发明的限制。
纳米uio-67-nh2的制备
实施例1
一种纳米uio-67-nh2的制备方法,包括如下步骤:
称量0.095g(0.37mmol)的2-氨基-4,4'-联苯二甲酸、0.067g(0.287mmol)的四氯化锆,分别将上述两种物质加入到7.5ml的dmf(n,n-二甲基甲酰胺)溶剂中,并超声分散处理;分散后,将上述分散所得两种物质混合搅拌20分钟,得到初混料;随后向初混料中加入1.4ml的冰醋酸和0.1ml的三乙胺,得到混合原料;然后将混合原料置于聚四氟乙烯内衬的高压反应釜中,在120℃下反应24小时,最后在100℃下真空干燥24小时,得到纳米uio-67-nh2。其粒径约为150nm,比表面积为240.14m2·g-1,孔容为415cm3·g-1。
实施例2
一种纳米uio-67-nh2的制备方法,包括如下步骤:
称量0.088g(0.36mmol)的2,2'-二吡啶-5,5'-二羧酸、0.125g(0.29mmol)的硝酸锆,分别将上述两种物质加入到7.7ml的n,n-二乙基甲酰胺溶剂中,并超声分散处理;分散后,将上述分散所得两种物质混合搅拌15分钟,得到初混料;随后向初混料中加入1.3ml的乙二胺,得到混合原料;然后将混合原料置于聚四氟乙烯内衬的高压反应釜中,在118℃下反应26小时,最后在102℃下真空干燥25小时,得到纳米uio-67-nh2。其粒径约为145nm,比表面积为250.14m2·g-1,孔容为425cm3·g-1。
实施例3
一种纳米uio-67-nh2的制备方法,包括如下步骤:
称量0.087g(0.34mmol)的2-氨基-4,4'-联苯二甲酸、0.058g(0.25mmol)的四氯化锆,分别将上述两种物质加入到7.6ml的dmf(n,n-二甲基甲酰胺)溶剂中,并超声分散处理;分散后,将上述分散所得两种物质混合搅拌25分钟,得到初混料;随后向初混料中加入1.5ml的冰醋酸和0.12ml的三乙胺,得到混合原料;然后将混合原料置于聚四氟乙烯内衬的高压反应釜中,在125℃下反应22小时,最后在105℃下真空干燥23小时,得到纳米uio-67-nh2。其粒径约为151nm,比表面积为240.22m2·g-1,孔容为414cm3·g-1。
实施例4
一种纳米uio-67-nh2的制备方法,包括如下步骤:
称量0.105g(0.41mmol)的2-氨基-4,4'-联苯二甲酸、0.058g(0.25mmol)的四氯化锆,分别将上述两种物质加入到8.0ml的dmf(n,n-二甲基甲酰胺)溶剂中,并超声分散处理;分散后,将上述分散所得两种物质混合搅拌25分钟得到初混料,随后向初混料中加入1.5ml的冰醋酸和0.1ml的三乙胺,得到混合原料;然后将所述混合原料置于反应釜中,在119℃下反应25小时,得到初产物,将所述初产物离心分离后,采用真空干燥方式,在102℃下真空干燥23.5小时,得到纳米uio-67-nh2。其粒径约为150nm,比表面积为240.34m2·g-1,孔容为417cm3·g-1。
实施例5
一种纳米uio-67-nh2的制备方法,包括如下步骤:
称量0.084g(0.325mmol)的2-氨基-4,4'-联苯二甲酸、0.061g(0.26mmol)的四氯化锆,分别将上述两种物质加入到9ml的dmf(n,n-二甲基甲酰胺)溶剂中,并超声分散处理;分散后,将上述分散所得两种物质混合搅拌22分钟,得到初混料;随后向初混料中加入1.44ml的冰醋酸和0.09ml的三乙胺,得到混合原料;然后将混合原料置于聚四氟乙烯内衬的高压反应釜中,在121℃下反应23小时,最后在103℃下真空干燥24.5小时,得到纳米uio-67-nh2。其粒径约为153nm,比表面积为241.34m2·g-1,孔容为413cm3·g-1。
纳米uio-67-nh2的理化性能检测
1.1、对实施例1中所得的纳米uio-67-nh2产品进行如下形貌和理化性能测试:
(1)、通过x-射线粉末衍射得到其晶体结构图(图1);
(2)、采用ultimaiv型x-射线粉末衍射仪检测(图2);
(3)、采用扫描电镜sem测试,采用supra55sapphire场发射扫描电子显微镜观测、评估其尺寸和形态(图3);
(4)、进行了bet分析,图4为n2吸附脱附等温线;图5为孔径分布曲线;
(5)、使用激光粒度分析仪(nano-zs马尔文粒径分布仪),进行粒径分布测试,评估其粒度分布(图6)。
1.2结果分析:
(1)由图1所示,图中的序号1为zr,2为o,3为c,4为nh2,图中结构可知,本发明所得的材料结构规则。
(2)由图2所示,纳米uio-67-nh2(图中nano-uio-67-nh2谱带)显示出与uio-67单晶模拟计算结果(图中simulation谱带)的一致的xrd图谱,表明本发明实施例1产品已经获得了uio-67-nh2的结构。
(3)由图3所示本发明实施例1产品纳米uio-67-nh2的纳米颗粒大部分呈八面体形状,少部分形状不规则;由图6所示本发明实施例1产品的平均粒径约为150nm,粒径呈正态分布,表明大多数粒子的尺寸小于200nm。
(4)由图4~5,表明本发明实施例1产品存在孔径为2~50nm的介孔。此外,曲线中的h4型滞回线揭示了圆柱形孔的存在。所述的孔径分布曲线图在2.05和3.85nm处显示出两个峰,这证实了在本发明实施例1产品中出现介孔,其比表面积为240.14m2·g-1,孔容为415cm3·g-1。较大的孔径可以载入一些尺寸较大的药物分子。
进一步的,对实施例2-5所制备得到的纳米uio-67-nh2进行与实施例1制备的纳米uio-67-nh2进行相同的表征,实施例2-5制备得到的纳米uio-67-nh2表征结果与实施例1高度吻合,说明了所制备的产品重现性极好。
纳米uio-67-nh2的应用
实施例6
一种纳米uio-67-nh2的应用方法:是将实施例1中所得的纳米uio-67-nh2用于装载药物,具体是将3mgcpt在10ml甲醇中超声溶解,之后将1mg纳米uio-67-nh2与加入到cpt溶液中,于温度为25℃条件下,搅拌装载7天。此时的载药率可达38.23w%,载药效率达到20.61%。
实施例7
一种纳米uio-67-nh2的应用方法:是将实施例3中所得的纳米uio-67-nh2用于装载药物,具体是将2.9mgcpt在10ml甲醇中超声溶解,之后将1mg纳米uio-67-nh2与加入到cpt溶液中,于温度为25℃条件下,搅拌装载7天。此时的载药率可达38.15w%,载药效率达到20.44%。
实施例8
一种纳米uio-67-nh2的应用方法:是将实施例4中所得的纳米uio-67-nh2用于装载药物,具体是将3mgcpt在10ml甲醇中超声溶解,之后将1.1mg纳米uio-67-nh2与加入到cpt溶液中,于温度为25℃条件下,搅拌装载7天。此时的载药率可达37.67w%,载药效率达到20.41%。
实施例9
一种纳米uio-67-nh2的应用方法:是将实施例5中所得的纳米uio-67-nh2用于装载药物,具体是将1mgcpt在10ml甲醇中超声溶解,之后将1mg纳米uio-67-nh2与加入到cpt溶液中,于温度为22℃条件下,搅拌装载6天。此时的载药率可达18.15w%,载药效率达到19.23%。
实施例10
一种纳米uio-67-nh2的应用方法:是将实施例2中所得的纳米uio-67-nh2用于装载药物,是将2mgcpt在10ml甲醇中超声溶解,之后将1mg纳米uio-67-nh2与加入到cpt溶液中,于温度为28℃条件下,搅拌装载5天。此时的载药率可达21.18w%,载药效率达到19.43%。
纳米uio-67-nh2的应用稳定性和红外光谱以及cpt释放性能检测
2.1、对实施例6加载药物后的纳米uio-67-nh2进行cpt释放性能和稳定性检测,具体是:
(1)进行zeta电位测量,以检测在载药前后uio-67-nh2纳米颗粒的电位变化;载药前zeta电位图见图7,载药后zeta电位图见图8;
(2)进行ft-ir红外光谱分析,分别对2-氨基-4,4'-联苯二甲酸(配体)、纳米uio-67-nh2、喜树碱(cpt)和载药后的uio-67-nh2纳米颗粒(纳米uio-67-nh2@cpt)进行了表征;图谱见图9;
图9中谱带由上至下依次表示:
ligand:2-氨基-4,4'-联苯二甲酸;
nano-uio-67-nh2:纳米uio-67-nh2;
cpt:喜树碱;
nano-uio-67-nh2@cpt:加载喜树碱后的纳米uio-67-nh2颗粒;
(3)进行xrd分析,分别对uio-67-nh2悬浮在磷酸盐缓冲溶液pbs(ph=7.4)中5天后,洗脱后的颗粒(nano-uio-67-nh2pbs)、悬浮在甲醇中5天后洗脱的颗粒(纳米uio-67-nh2)、在甲醇中加载喜树碱后的uio-67-nh2纳米颗粒(cpt@nano-uio-67-nh2)进行了xrd表征;图谱见图10;
图10中谱带由上至下依次表示:
nano-uio-67-nh2:悬浮在甲醇中5天后洗脱的颗粒纳米uio-67-nh2;
cpt@nano-uio-67-nh2:甲醇中加载喜树碱后的纳米uio-67-nh2颗粒;
nano-uio-67-nh2pbs:纳米uio-67-nh2在磷酸盐缓冲水溶液中洗脱后的颗粒;
(4)将加载喜树碱后的纳米uio-67-nh2(cpt@nano-uio-67-nh2)置于透析袋中,然后将其浸入pbs(磷酸盐缓冲溶液ph=7.4)中,于37℃搅拌7天,用于持续的药物释放研究。通过高速液相色谱(hplc)测量pbs中的喜树碱(cpt)浓度;见图11。
2.2结果分析:
(1)由图7~8所示,载药前后uio-67-nh2纳米颗粒显示zeta电位分别显示-12.5和-14.3mv,这些zeta电位数据位于正常范围内,表明纳米uio-67-nh2在装载药物后是稳定的。
(2)由图9所示,纳米uio-67-nh2@cpt光谱的1744cm-1处吸收带,归属于cpt内酯环的c=o的伸缩振动。在1437、1412cm-1的两个带,分别归属于cpt的o-h弯曲振动和纳米uio-67-nh2的n-h弯曲振动。在1156cm-1处归属于cpt内酯环的c-o-c伸缩振动。在cpt加载后,cpt的特征带与纳米uio-67-nh2一起出现,表明载体上存在cpt。
(3)由图10所示,nano-uio-67-nh2与cpt@nano-uio-67-nh2和通过uio-67-nh2单晶模拟计算的xrd曲线没有显著差异,表明纳米uio-67-nh2以及负载了cpt的uio-67-nh2在甲醇中是稳定的。将uio-67-nh2悬浮在磷酸盐缓冲溶液pbs(ph=7.4)中5天后,nano-uio-67-nh2与nano-uio-67-nh2pbs通过uio-67-nh2单晶模拟计算的xrd曲线没有显著差异,说明nano-uio-67-nh2在磷酸盐缓冲水溶液也是稳定的;这表明在4,4'-联苯二甲酸上修饰了氨基后,提高了mof的水稳定性。
(4)由图11所示,通过hplc测量cpt浓度。约有74%的cpt在5天内释放。cpt释放分为三个阶段:依次为第0~22、22~96和96~120小时;在第一、第二和第三阶段,分别释放45%、25%和4%的药物。实验结果表明,在释放过程中没有发生“药物突释”,并且实现了持续的药物释放。
进一步的,对实施例7-10加载药物后的纳米uio-67-nh2进行了与实施例6一致的负载性能和稳定性检测,实施例7-10检测所得的结果与实施例6高度吻合,说明了所制备的产品稳定性极好。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种纳米粉体工业生产系统的制作方法
- 一种高红外辐射粉体及其制备方法
- 一种铈锌共掺NiFe<sub>2</sub>O<sub>4</sub>纳米粉体及其制备方法
- 一种纳米Al<sub>2</sub>O<sub>3</sub>/Er<sub>3</sub>Al<sub>5</sub>O<sub>12</sub>/ZrO<sub>2</sub>复合粉体材料的制备方法
- 一种β?氢氧化镍纳米片的制备方法
- 一种硫化亚铜纳米玫瑰花的制备方法
- 一种球形氧化钇粉体的制备方法
- 一种亚微米级碳化硼粉体及其制备方法和用图
- 一种B<sub>4</sub>C纳米片的制备方法及B<sub>4</sub>C纳米片的制作方法
- 氮化铝粉体的制备方法