从知母中分离的具有抗肿瘤活性的化合物及其制备方法与流程
本发明涉及从植物中分离得到的具有抗肿瘤活性的化合物,尤其涉及从知母须根中分离得到的具有抗肝癌活性的化合物,属于抗癌化合物领域。
背景技术:
中药知母为百合科植物知母anemarrhenaasphodeloidesbge.的干燥根茎,其主要分布于中国、蒙古等东亚地区,广泛用于治疗各种疾病,如发热、糖尿病等。知母须根作为非药用部位,在知母加工过程中常被大量舍弃,这不仅造成了资源浪费,也加重了环境污染。知母须根的化学成分和药理作用与知母基本相同。因此,开发利用知母须根资源是有必要的。
肝细胞性肝癌(hepatocellularcarcinoma)对全球公共卫生产生重大影响。化疗是现在的主要治疗方法,但其非选择性也导致正常细胞死亡。现有的治疗肝细胞性肝癌的化学药物均存在疗效不够显著的缺陷,从植物中分离得到新的具有显著抗肝癌活性的化合物,这对于肝癌的治疗具有非常重要的意义。
技术实现要素:
本发明目的之一是提供一种从知母须根分离得到的具有抗肿瘤活性的化合物。
本发明的目的之二是提供一种从知母须根中分离得到该化合物的方法。
本发明的目的之三是将该化合物制备成适用于临床应用的各种抗肿瘤制剂。
本发明的上述目的是通过以下技术方案来实现的:
从知母须根中分离得到的具有抗肿瘤活性的化合物,其化学结构为为式ⅰ、式ⅱ、式ⅲ以及式ⅳ所示:
其中,r1=氢或oh,r2=氢或oh,r3=oh或och3,r4=h或ch2cooch3,r5=氢或oh,r6=氢或oh,r7=氢或oh,r8=氢或oh,r9=氢或oh,r10=氢或oh;
其中,式ⅱ化合物的分子式为c20h20o8,呈黄色油状,其英文名称全(2,6-dihydroxy-3-(((1s,4s,6r)-4-hydroxy-3,7-dioxabicyclo[4.1.0]heptan-4-yl)methyl)-4-methoxyphenyl)(4-hydroxyphenyl)methanone。
进一步优选的,所述的式ⅰ化合物是式ⅴ或式ⅵ所示结构的化合物:
上述式ⅰ、式ⅱ、式ⅲ以及式ⅳ化合物的酸加成的盐也包括在本发明之中。
所述酸加成的盐优选为药学上可以接受的与适当的酸(例如盐酸、醋酸、硫酸)形成无毒的盐,除了药物上可接受的盐以外,其它的盐也包括在本发明之中。
本发明进一步提供了一种从知母须根中分离得到式ⅱ所述化合物的方法,包括:
(1)将干燥的知母须根粉碎并用乙醇渗漉提取;
(2)将乙醇渗漉液进行减压浓缩得到粗提物;
(3)将粗提物用水分散后用有机溶剂进行萃取,将萃取后剩余下的水部位用水分散,溶液流经大孔树脂柱用乙醇-水进行洗脱;收集90%乙醇部分经硅胶柱纯化后用二氯甲烷-甲醇洗脱,依次得到七个组分,即:fr.1、fr.2、fr.3、fr.4、fr.5、fr.6和fr.7;
(4)将用二氯甲烷-甲醇=100:1洗脱得到的fr.1组分进行硅胶柱洗脱,洗脱条件是用石油醚-乙酸乙酯系统梯度洗脱20:1~1:1,得到fr.1.1、fr.1.2、fr.1.3和fr.1.4四个组分;将用石油醚-乙酸乙酯=5:1洗脱得到的fr.1.3组分经制备柱色谱得到化合物4(式ⅵ化合物);
(5)将用二氯甲烷-甲醇=20:1进行洗脱所得到的fr.2通过ods柱,依次用30%甲醇,60%甲醇和90%甲醇洗脱,依次获得8个流分,即:fr.2.1-fr.2.8;其中,用90%甲醇洗脱部分得到的fr.2.8用二氯甲烷:甲醇按照100:1→0:1的洗脱程序在硅胶柱上洗脱,依次得到8个洗脱组分,即:fr.2.8.1-fr.2.8.8;将其中用二氯甲烷-甲醇=50:1的洗脱条件得到的fr.2.8.1进行制备型柱色谱(甲醇-水=55:45,7ml·min-1)得到fr.2.8.1.1-fr.2.8.1.4;将fr.2.8.1.4经半制备柱色谱得到化合物2(式ⅴ);
(6)将用二氯甲烷-甲醇=20:1洗脱条件洗脱得到的fr.2.8.4进行制备型柱色谱(甲醇-水=54:46,流速是8ml·min-1)得到fr.2.8.4.1-fr.2.8.4.6;将fr.2.8.4.2用半制备柱色谱法得到化合物1(式ⅱ);
(7)将用90%甲醇洗脱得到的fr.2.7进行硅胶柱洗脱,用流动相二氯甲烷-甲醇按照从100:1~1:1进行洗脱,得到fr.2.7.1~fr.2.7.4;将用二氯甲烷-甲醇=10:1的洗脱条件洗脱得到的fr.2.7.3经制备柱色谱(甲醇-水=48:52,流速是7ml·min-1)依次得到fr.2.7.3.1~fr.2.7.3.4,再将fr.2.7.3.4经半制备柱色谱得到化合物5(式ⅳ);
(8)将用60%甲醇洗脱得到的fr.2.6进行硅胶柱洗脱,用流动相二氯甲烷-甲醇按照从50:1~1:1的洗脱梯度进行洗脱得到fr.2.6.1~fr.2.6.4,将用二氯甲烷-甲醇=20:1的洗脱条件洗脱得到的fr.2.6.2经制备柱色谱(流动相是甲醇-水=39:61,洗脱流速是7ml·min-1)得到fr.2.6.2.1~fr.2.6.2.6,再将fr.2.6.2.5经半制备柱色谱(tr=45.3min,乙腈-水=25:75)得到化合物3(式ⅲ)。
优选的,步骤(1)中所述的乙醇优选为40-95乙醇,优选为70%乙醇。
优选的,步骤(3)中将粗提物用水分散后用乙酸乙酯进行萃取;步骤(3)中所述的大孔树脂柱优选为ab-8大孔树脂柱;步骤(3)中用10:90~90:10的乙醇-水进行洗脱。步骤(3)中所述的硅胶柱优选为200~300目的硅胶柱;步骤(3)中所述的二氯甲烷-甲醇的体积配比为100:1~0:1。
本发明采用制备液相色谱设备与材料,所用材料均可重偶复利用,大大降低了对环境的污染。
对本发明化合物进行体外抗肝癌试验,试验结果表明本发明化合物具有显著的抑制肝癌的效果。
所述的肿瘤包括肝癌、胃癌、肺癌、肾癌、结肠癌等,优选为肝癌。
本发明的另外一个目的是提供一种抗肿瘤的药物组合物,该药物组合物由治疗上有效量的化合物或其可药用的盐与药学上可接受的载体配合而成;
将药学上可接受用量的化合物与药学上可接受的载体或稀释剂配合后,按本领域常规的制剂方法将其制备成任意一种适宜的药物组合物。通常该组合物适合于口服给药和注射给药,也适合其他的给药方法。该组合物可以是片剂、胶囊剂、粉剂、颗粒剂、锭剂、栓剂,或口服液等液体制剂形式。根据不同的给药方法,本发明药物组合物可以含有0.1%-99%重量,优选10-60%重量的单体化合物。
所述的载体是指药学领域常规的载体,例如:稀释剂、赋形剂如水等;粘合剂可以是纤维素衍生物、明胶、聚乙烯吡咯烷酮等;填充剂可以是淀粉等物质;崩裂剂可以是碳酸钙或碳酸氢钙等;另外,还可以药物组合物加入其它辅助剂如香味剂和甜味剂。
按照常规的制剂工艺,可以将本发明所述的抗肿瘤的药物组合物制成口服制剂或注射药物制剂。所述的口服制剂可以是片剂、丸剂、散剂、膏剂、胶囊剂、口服液或颗粒剂等;所述的注射制剂可以是粉针剂或注射液等。
本发明药物制剂的施用量可根据用药途径、患者年龄、体重、所治疗的肿瘤类型和严重程度等进行相应的变化,这些都是本领域人员所习知。
本发明药物制剂可通过临床可接受的治疗方式给药,包括注射、口服、吸入、植入或灌注等。
附图说明
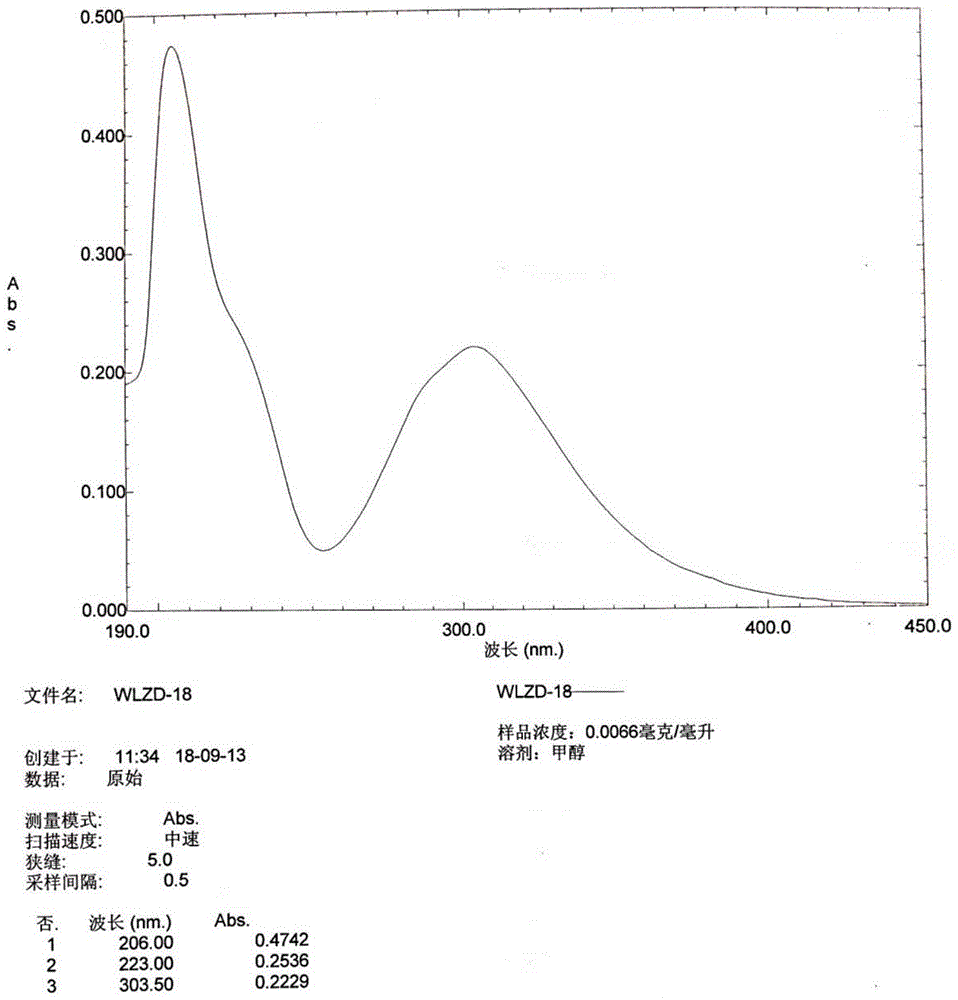
图1化合物1的uv波谱图;
图2化合物1的ir波谱图;
图3化合物11hnmr的波谱图(500mhz,cd3od);
图4化合物113cnmr的波谱图(125mhz,cd3od);
图5化合物1dept波谱图(125mhz,cd3od);
图6化合物1hmbc的波谱图(500mhz,cd3od);
图7化合物1hsqc的波谱图(500mhz,cd3od);
图8化合物11h-1hcosy的波谱图(500mhz,cd3od);
图9化合物1hresims的波谱图(500mhz,cd3od);
图10化合物2的uv波谱图;
图11化合物2的ir波谱图;
图12化合物21hnmr的波谱图(500mhz,cd3od);
图13化合物213cnmr的波谱图(125mhz,cd3od);
图14化合物2hmbc的波谱图(500mhz,cd3od);
图15化合物2hsqc的波谱图(500mhz,cd3od);
图16化合物21h-1hcosy的波谱图(500mhz,cd3od);
图17化合物2hresims的波谱图(500mhz,cd3od);
图18化合物31hnmr的波谱图(600mhz,dmso-d6);
图19化合物313cnmr的波谱图(150mhz,dmso-d6);
图20化合物41hnmr的波谱图(600mhz,dmso-d6);
图21化合物413cnmr的波谱图(150mhz,dmso-d6);
图22化合物41hnmr的波谱图(800mhz,dmso-d6);
图23化合物513cnmr的波谱图(200mhz,dmso-d6);
图24化合物5dept波谱图(200mhz,dmso-d6);
图25化合物5hsqc的波谱图(800mhz,dmso-d6);
图26化合物5hmbc的波谱图(800mhz,dmso-d6)。
具体实施方式
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但是应理解所述实施例仅是范例性的,不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改或替换均落入本发明的保护范围。
实施例1抗肿瘤化合物的分离制备及鉴别
将干燥的知母须根(30kg)粉碎并用70%乙醇渗漉提取。减压浓缩乙醇渗漉液,得到棕色粗提物。将粗提物用水分散并用乙酸乙酯萃取。将萃取后剩余下的水部位用水分散,溶液流经ab-8大孔树脂柱,用乙醇-水洗脱(10:90和90:10),90%乙醇部分经硅胶柱(200~300目)进一步纯化,用二氯甲烷-甲醇(100:1~0:1)洗脱,得到fr.1~fr.7。将fr.1(二氯甲烷-甲醇=100:1部分)进行硅胶柱洗脱(用石油醚-乙酸乙酯系统梯度洗脱20:1~1:1)得到fr.1.1~fr.1.4。fr.1.3(石油醚-乙酸乙酯=5:1部分)经制备柱色谱得到化合物4(7mg,tr=20.3min,甲醇-水=51:49)。fr.2(二氯甲烷-甲醇=20:1部分)通过ods柱(依次用30%甲醇,60%甲醇和90%甲醇洗脱)获得8个流分fr.2.1-fr.2.8。fr.2.8(90%甲醇洗脱部分)用二氯甲烷:甲醇(100:1→0:1)在硅胶柱上洗脱,得到fr.2.8.1-fr.2.8.8。将fr.2.8.1(二氯甲烷-甲醇=50:1部分)进行制备型柱色谱(甲醇-水=55:45,7ml·min-1),得到fr.2.8.1.1-fr.2.8.1.4。将fr.2.8.1.4经半制备柱色谱得到化合物2(8mg,tr=35.3min,乙腈-水=36:64)。将fr.2.8.4(二氯甲烷-甲醇=20:1部分)进行制备型柱色谱(甲醇-水=54:46,8ml·min-1),得到fr.2.8.4.1-fr.2.8.4.6。fr.2.8.4.2用半制备柱色谱法得到化合物1(tr=22.4min,乙腈-水=32:68)。fr.2.7(90%甲醇洗脱部分)进行硅胶柱洗脱,用流动相二氯甲烷-甲醇(从100:1~1:1洗脱)得到fr.2.7.1~fr.2.7.4。将fr.2.7.3(二氯甲烷-甲醇=10:1部分)经制备柱色谱(甲醇-水=48:52,7ml·min-1)得到fr.2.7.3.1~fr.2.7.3.4,再将fr.2.7.3.4经半制备柱色谱得到化合物5(23mg,tr=59.1min,乙腈-水=26:74)。fr.2.6(60%甲醇洗脱部分)进行硅胶柱洗脱,用流动相二氯甲烷-甲醇(从50:1~1:1洗脱)得到fr.2.6.1~fr.2.6.4。将fr.2.6.2(二氯甲烷-甲醇=20:1部分)经制备柱色谱(甲醇-水=39:61,7ml·min-1)得到fr.2.6.2.1~fr.2.6.2.6,再将fr.2.6.2.5经半制备柱色谱得到化合物3(10mg,tr=45.3min,乙腈-水=25:75)。
化合物1(式ⅱ)为黄色油状,
化合物1的鉴别
氢、碳谱数据如下:13cnmr(125mhz,cd3od).δc198.1(c-7),165.4(c-2),162.8(c-4),162.4(c-4′),160.2(c-6),133.0(c-1′),132.2(c-2′,6′),115.5(c-3′,5′),114.2(c-6″),105.0(c-5),103.3(c-1),93.5(c-3),68.2(c-3″),67.3(c-2″),66.7(c-4″),56.3(4-och3),39.9(c-7″),37.7(c-5″);1hnmr(500mhz,cd3od),δh7.51(2h,d,j=8.7hz,h-2′,6′),6.81(2h,d,j=8.7hz,h-3′,5′),6.13(1h,s,h-3),3.84(3h,s,4-och3),3.60(2h,m,h-2″),3.52(1h,m,h-3″),3.48(1h,m,h-4″),2.97(1h,d,j=16.0hz,h-7″),2.90(1h,d,j=16.0hz,h-7″),2.06(1h,t,j=12.4hz,h-5″),1.89(1h,dd,j=12.9,4.7hz,h-5″).
化合物1的各种波谱鉴别见图1-图9。
化合物2的鉴别
红棕色无定型粉末(甲醇);hr-esi-ms给出准分子离子m/z355.0733[m+na]+(calcdfor355.0794)。uv(meoh)λmax(logε):204(4.59),224(4.30),303(4.13)nm。
化合物2的氢、碳谱数据如下:1hnmr(500mhz,cd3od):δh7.58(2h,d,j=8.8hz,h-2′,-6′),δh6.76(2h,d,j=8.8hz,h-3′,-5′),δh6.05(1h,s,h-3),δh3.58(2h,s,h-1”),δh3.81(3h,s,4-och3),δh3.65(3h,s,2”-och3)。13cnmr(500mhz,cd3od):133.2(c-1′),132.9(c-2′,6′),115.4(c-3′,5′),161.1(c-4′),106.8(c-1),161.1(c-6),102.8(c-2),164.3(c-3),91.8(c-4),160.8(c-5),199.3(c-7),28.7(c-1″),175.0(c-2″),56.2(4-och3),52.4(2″-och3)。
化合物2的各种波谱鉴别见图10-图17。
化合物3的鉴别
黄色油状(甲醇);1hnmr(600mhz,dmso-d6):δh7.59(d,2h,j=8.7hz,h-2′,-6′),δh6.79(d,2h,j=8.7hz,h-3′,-5′),δh6.13(s,1h,h-5),δh3.91(d,1h,j=5.8hz,h-3″),δh3.63(m,1h,h-4″),δh3.56(m,1h,h-5″),δh3.53(m,1h,h-6″),δh3.37(m,1h,h-6″),δh3.31(d,1h,j=16.3hz,h-1″),δh2.67(d,1h,j=16.3hz,h-1″),δh3.79(s,3h,och3);13cnmr(125mhz,dmso-d6):δc103.2(c-1),157.9(c-2),103.5(c-3),157.1(c-4),92.2(c-5),158.8(c-6),192.2(c-7),129.1(c-1′),132.0(c-2′),114.9(c-3′),162.3(c-4′),114.9(c-5′),132.0(c-6′),32.2(c-1″),120.0(c-2″),80.8(c-3″),75.8(c-4″),82.0(c-5″),60.6(c-6″),55.5(och3)。
化合物3的各种波谱鉴别见图18-图19。
化合物4的鉴别
无色结晶(甲醇);1hnmr(600mhz,dmso-d6):δh7.55(d,2h,j=8.5hz,h-2′,-6′),δh6.88(d,2h,j=8.5hz,h-3′,-5′),δh6.96(s,1h,j=8.7hz,h-6),δh6.40(s,1h,j=8.7hz,h-5);13cnmr(125mhz,dmso-d6):δc113.1(c-1),151.9(c-2),151.9(c-3),132.7(c-4),107.4(c-5),124.5(c-6),197.9(c-7),128.9(c-1′),131.6(c-2′),115.0(c-3′),161.0(c-4′),115.0(c-5′),131.6(c-6′)。
化合物4的各种波谱鉴别见图20-图22。
化合物5的鉴别
绿色无定型粉末(甲醇);hr-esi-ms给出准分子离子m/z366.0947[m+na]+(calcdfor366.0954)。
化合物5的氢、碳谱数据如下:1hnmr(800mhz,dmso-d6):δh7.53(2h,d,j=8.7hz,h-2′,-6′),δh7.44(1h,s,nh),δh6.78(2h,d,j=8.7hz,h-3′,-5′),δh6.08(1h,s,h-3),δh5.11(1h,d,j=4.9,9.3hz,h-1”),δh3.74(3h,s,4-och3),δh2.32(1h,m,h-3”),δh2.28(1h,m,h-2”),δh2.16(1h,m,h-3”),δh2.01(1h,m,h-2”);13cnmr(200mhz,dmso-d6):130.8(c-1′),131.6(c-2′,6′),114.6(c-3′,5′),161.8(c-4′),106.8(c-1),158.3(c-2),91.5(c-3),161.5(c-4),108.6(c-5),157.8(c-6),195.8(c-7),46.9(c-1″),25.8(c-2″),30.7(c-3″),176.8(c-4″),55.5(4-och3)。
化合物5的各种波谱鉴别见图23-图26。
试验例1本发明化合物体外抗肿瘤活性试验
本试验中所用的细胞株为国际通用的肝癌细胞株—肝癌细胞hepg2、hep3b。
试验方法采用国际通用的mtt测定方法,具体如下:
将对数生长期细胞以1×105cells/孔接种于96孔板,置于37℃,5%co2条件下培养直至细胞90%融合后,用无血清培养基孵育2h使细胞同步化。随后,弃去上清,分别加入含有各化合物(实施例1所制备)(10nm,100nm,1000nm,5000nm,10000nm)的emem培养基孵育72h,孵育结束前4h,每孔加入20μlmtt溶液(5mg/ml)。孵育结束后,弃去各孔上清液,每孔加入150μldmso,细胞振荡仪上振荡10min,待结晶物充分溶解后用酶标仪测定od570。
测定结果发现,本发明化合物均具有显著的体外抗肝癌活性。
表1不同的化合物对于hepg2和hep3b细胞的生长抑制试验结果(ic50valuesinnmol/l)
注:与5-fu相比,*p<0.05,**p<0.01
- 还没有人留言评论。精彩留言会获得点赞!