一种寡核苷酸组、一种核酸序列的扩增方法及目的蛋白的表达方法与流程
本发明属于生物技术领域,具体涉及一种寡核苷酸组、一种核酸序列的扩增方法及目的蛋白的表达方法。
背景技术:
作为遗传信息载体的脱氧核糖核酸序列含有很多种基因,其涉及到调节和控制生物体的生长和发育。随着核酸检测技术的发展,人们发现dna寡核苷酸与人类疾病密切相关,因此,核酸序列检测方法的高特异性和灵敏性在医疗诊断、司法鉴定以及环境和食品安全监督中起着重要的作用。
为了满足医疗诊断、司法鉴定等方面的需要,现已经有一些核酸扩增策略被设计用于核酸序列的检测。比如聚合酶链式反应(pcr),它提供了很高的分析灵敏度,pcr技术在对于依赖检测和定量的dna靶标的医学分子诊断学中掀起了一场革命性的变革。pcr在遗传分析、dna克隆、体外诊断、传染病快速筛查等方面有很好的效果。滚环扩增(rca)通常也被用于核酸序列的检测,它是新近发展起来的一种恒温核酸扩增方法。rca以环状dna为模板,通过一个短的dna引物(与部分环状模板互补),在酶催化下将dntps转变成单链dna,此单链dna包含成百上千个重复的模板互补片段,还可以实现对靶核酸的信号放大,因此在核酸检测中具有很大的应用价值和潜力。环介导的等温扩增(lamp)是一种恒温扩增反应,其特征在于使用六个专门设计的引物区域来识别靶dna上的八个区域,基因的扩增和检测可以通过在60至65℃的等温条件反应物一起温育而一步完成,因此特异性较高。
然而pcr方法,因为需要精确的温度和循环控制,在环境条件较差的地方,因为条件不足难以广泛的推广;滚环扩增技术这些方法基于模板复制,增加了扩增子交叉污染的风险并导致频繁发生假阳性结果;环介导的等温扩增需要很精确的引物设计,因此容易造成假阳性的效果出现。
技术实现要素:
有鉴于此,本发明的目的在于针对现有技术的缺陷,提供一种操作简单、结果准确率高的检测核酸序列的方法。
为实现本发明的目的,本发明采用如下技术方案:
本发明提供了一种寡核苷酸组,由probe和trigger组成;
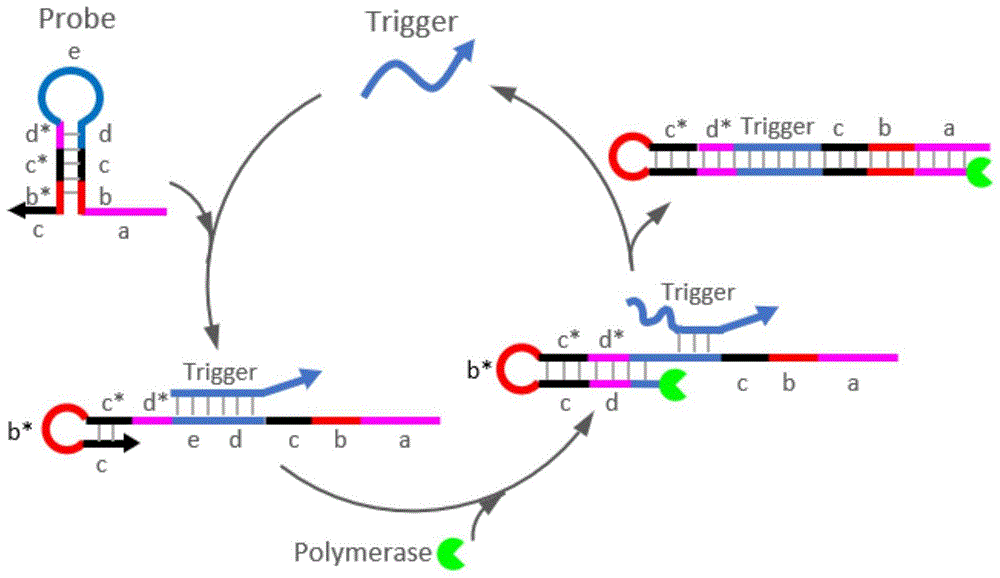
所述probe由依次连接的下述五个部分组成:第一部分为a区域,第二个部分由b、c、d三个区域组成,第三个部分为e区域,第四个部分由b*、c*、d*三个区域组成,第五个部分为c区域;其中第二部分的b、c、d三个区域与第四部分的b*、c*、d*三个区域分别反向互补配对;且a区域与c区域不能互补配对,e区域既不可与d区域碱基互补也不可与d*区域碱基互补;e和d碱基数之和大于b、c、d区域的碱基数之和;
所述trigger由依次连接的d*区域、e*区域和3个以上的胸腺嘧啶组成。
本发明所述寡核苷酸组中的probe为发夹结构;trigger为短的寡核苷酸序列,可作为触发器。probe和trigger在温育中,trigger会作为触发器,trigger序列中的e*和d*区域会分别和probe序列中的e和d区域进行互补配对,由于e和d碱基数之和大于b、c、d区域的碱基数之和,因此trigger会打开probe的第二部分与第四部分之间的碱基配对,形成以c区域为引物的二级结构(可参照附图)。在形成这个复杂的二级结构之后,当加入具有链取代功能的dna聚合酶时,c区域会作为这个复杂二级结构的引物以c*d*edcba这条链为模板进行序列延伸;当延伸到d*末端时,dna聚合酶就会发挥它的链取代活性,将e*和d*取代下来继续作为trigger加入到下一轮反应中继续打开probe探针,同时在dna聚合酶作用下继续向前滑动进行序列延伸,实现trigger在反应时被多次循环重复利用的效果。
所述trigger序列中的d*碱基末端连续的胸腺嘧啶序列是为了防止在dna聚合酶的作用下进行扩增,因此个数没有特别的限制,只要可以满足该序列不被延伸即可。在一些实施方案中,所述trigger序列中的胸腺嘧啶个数为6个。
在一些实施方案中,所述probe的序列如seqidno.1所示,所述trigger的序列如seqidno.2所示。
在一些实施方案中,所述寡核苷酸组中的probe中的a区域为依次连接的切口酶识别位点序列和g-四连体序列。当聚合酶将变形后的probe探针修补为双链之后,加入切口酶会在a*区打开切口,在dna聚合酶的作用下切口处不断地进行链取代扩增,产生大量的g-四连体序列,将信号扩大,再加入硫黄素t(tht)之后,产生荧光,同时达到能够准确地检测目的序列的效果。
作为优选的,所述的寡核苷酸组中所述probe的序列如seqidno.3所示,所述trigger的序列如seqidno.2所示。
在一些实施方案中,所述的寡核苷酸组中的probe中的所述a区域为lacz基因序列。当dna聚合酶将变形后的probe探针修补为双链之后,将其放入到cell-free反应系统中进行反应,然后根据反应吸光度来检测反应的变化情况。
作为优选,所述的寡核苷酸组中所述probe的序列如seqidno.4所示,所述trigger的序列如seqidno.2所示。
本发明还提供了一种核酸序列的扩增方法,将所述的寡核苷酸组中的probe和te缓冲液混合变性后与所述的寡核苷酸组中的trigger37℃温育,然后加入dna聚合酶进行扩增反应;其中所述寡核苷酸组中的probe的a区域为目标核酸序列。
作为优选,所述的扩增方法中所述扩增反应的反应体系为1×blue缓冲液、0.33u/μlklenowfragmentexo-和250μmdntps。
本发明还提供了一种目的蛋白的表达方法,将所述的寡核苷酸组中的probe和te缓冲液混合变性后与所述的寡核苷酸组中的trigger37℃温育,之后加入dna聚合酶进行扩增反应,然后将扩增产物放入到cell-free反应系统中进行表达;其中所述寡核苷酸组中的probe的a区域为编码目的蛋白的核酸序列和rbs位点。
在probe经过变性之后,在聚合酶的作用下,形成由单链dna组成的双链结构(此时的rbs位点以及laczα基因均为双链结构),然后放入无细胞蛋白系统中,经过转录和翻译,表达laczα蛋白;由于无细胞蛋白系统中含有laczδm15蛋白,它与laczα蛋白可以相互结合,具有β-半乳糖苷酶的活性,通过与o-硝基苯-β-d-吡喃半乳糖苷(onpg)结合,从而呈现吸光值的变化。
由上述技术方案可知,本发明提供了一种寡核苷酸组、核酸序列的扩增方法及目的蛋白的表达方法。本发明与现有的dna基因检测相比有显著的进步,其主要优点如下:(1)可以在条件较差资源不足的地方应用,不需要专门的设备。在本发明中我们采用的温育方式包括二级结构的形成以及序列的延伸基本上均是恒温37℃,不需要像pcr有着特定的温度变化要求,同时也省去了退火温度的优化与选择,应用起来更加广泛。(2)实验过程操作快捷方便。整个实验操作过程中只需要添加病毒核酸dna以及检测探针probe,该probe序列经过设计在nupack中一分钟内就能够验证所设计的序列能否得到所需结构。(3)检测的灵敏度相对于其他实验来说更加准确。本发明的trigger可以循环利用,在检测dna核酸时,即在微量的dna核酸情况下,也可以准确且快速的检测。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
图1示实施例1荧光检测原理图;
图2示实施例1聚丙烯酰氨凝胶电泳检测反应的效果图;其中泳带1表示单独加入probe(100nm);泳带2表示单独加入probe(1μm);泳带3表示trigger(100nm)+probe(100nm);泳带4表示trigger(1μm)+probe(100nm);泳带5表示trigger(100nm)+probe(1μm);泳带6表示trigger(100nm)+probe(100nm)+klenowfragmentexo-dna聚合酶;泳带7表示trigger(1μm)+probe(100nm)+klenowfragmentexo-dna聚合酶;泳带8表示trigger(100nm)+probe(1μm)+klenowfragmentexo-dna聚合酶;
图3示实施例2荧光检测原理图;
图4示实施例2荧光检测图;横坐标代表波长,纵坐标代表荧光值,曲线排列从低到高依次是:probe-g+trigger;probe-g+trigger+bst2.0dna聚合酶;probe-g+bst2.0dna聚合酶+nt.bst.nbi切口酶;probe-g+trigger+bst2.0dna聚合酶+nt.bst.nbi切口酶;其中probe-g+trigger与probe-g+trigger+bst2.0dna聚合酶两组数据是重叠的;
图5示实施例2trigger浓度极限检测结果图;图中从下至上trigger浓度依次是0nm、1nm、5nm、10nm、20nm、50nm、100nm、150nm、1500nm;
图6示实施例3控制蛋白表达的原理图;
图7示实施例3控制蛋白表达的检测结果图;其中,横坐标表示时间,纵坐标表示吸光值。
具体实施方式
本发明公开了一种检测核酸序列的方法以及一种检测核酸序列的寡核苷酸序列。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及产品已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法进行改动或适当变更与组合,来实现和应用本发明技术。
为了进一步理解本发明,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
如无特殊说明,本发明实施例中所涉及的试剂均为市售产品,均可以通过商业渠道购买获得。其中合成的所有寡核苷酸序列是从genewiz(中国苏州)获得。bst2.0dna聚合酶和nt.bst.nbi切口酶购自newenglandinc.(ipswich,unitedstates)。klenowfragmentexo购自诺唯赞(中国南京)。2-[4-(二甲基氨基)苯基]-3,6-二甲基苯并噻唑氯化物(硫磺素t,tht)、o-硝基苯-β-d-吡喃半乳糖苷(onpg)和dnase/rnase-freeddh2o购自索莱宝(中国北京)。easytaqdna聚合酶和dntps购自全式金(中国北京)。sybrgold购自(invitrogen,usa)。温育和pcr实验均在pcr仪(eppendorf,germany)上进行,在离心机(thermofisherscientific,unitedstates)上进行离心实验。通过microplate读数器(thermofisherscientific,unitedstates)测量lacz蛋白和onpg在无细胞系统中的吸收光谱。用microplate读数器(tecan,switzerland)进行荧光光谱数据。使用c300成像系统(azurebiosystems,usa)记录凝胶图像。各实施例中序列信息如表1所示。
表1序列信息
实施例1、可行性分析
为了验证该反应原理的可行性进行了聚丙烯酰氨凝胶电泳来检查反应的效果。
实验方法:将100nm的probe和1×te缓冲液(tris-edta)混合,然后将混合物在95℃变性3分钟并缓慢冷却至室温。之后,将trigger加入反应物中并在37℃下孵育30分钟,然后加入1×blue缓冲液(10mmtris-hclph7.9,50mmnacl,10mmmgcl2,1mmdtt)、0.33u/μlklenowfragmentexo-(缺失3’→5’外切酶活性的klenow片段)和250μmdntps,在37℃温育30分钟终止反应。使用15%非变性聚丙烯酰胺凝胶电泳(page)分析产物。在1×tbe缓冲液(tris-硼酸盐-edta)中100v下电泳4小时。sybrgold用于在室温下染色凝胶。使用c300成像系统记录凝胶图像,结果图2所示。
图2结果显示泳道1和2是probe的片段作为对照,泳道3、4和5是在probe的基础上加入了trigger片段,根据互补配对的原理形成了新的结构(图2框内条带);在泳道3和4框的下方也是没有与trigger结合的probe的剩余量,发现当加大trigger的量时(100nm-1μm),剩余量减少,条带变暗;当加入dna聚合酶之后,如泳道6和7,不再有probe的剩余量条带出现,加入的所有probe在dna聚合酶的作用下进行了延伸,对于泳道8虽然probe条带并没有完全消失,但是相对于泳道2和5来说,条带亮度暗了很多;如果没有循环往复的效果,那么泳道6和7的probe条带位置就会有像泳道3和4一样亮度的条带出现,而实验结果中可以明显的发现,该位置并没有条带出现,所以也说明反应出现了循环往复利用的效果,同时也验证了整个反应的可行性。
实施例2、dna传感器的荧光检测
反应原理如图3所示,序列设计仍采用之前的序列设计方法,但是将a区的随机序列改为切口酶识别位点序列和g-四连体序列。当聚合酶将变形后的probe探针修补为双链之后,加入切口酶会在a*区打开切口,在dna聚合酶的作用下切口处不断地进行链取代扩增,产生大量的g-四连体序列,将信号扩大,再加入硫黄素t(tht)之后,产生荧光,同时达到能够准确地检测目的序列的效果。
实验方法:将150nm的probe和1×te缓冲液(tris-edta)混合,然后将混合物在95℃变性3分钟并缓慢冷却至室温。之后,将1.5μmtrigger加入混合物中并在37℃下孵育30分钟。接下来,将反应物中加入1×isothermalamplification缓冲液、0.5×nebuffertm3.1、0.32u/μlbst2.0dna聚合酶、0.4u/μlnt.bst.nbi、250μmdntps、6mmmgso4在55℃温育30分钟。最后,将产物在50mmtris-hcl、50mmkcl和7.5μmtht中温育20分钟。用酶标仪进行荧光光谱数据获得,结果如图4。
图4结果可见,曲线排列从低到高依次是:probe-g+trigger;probe-g+trigger+bst2.0dna聚合酶;probe-g+bst2.0dna聚合酶+nt.bst.nbi切口酶;probe-g+trigger+bst2.0dna聚合酶+nt.bst.nbi切口酶。表明荧光检测实验结果有着很高的特异性。
按照上述方法检测分别加入不同浓度的trigger检测的trigger加入的极限值(图5),结果显示所述荧光检测trigger浓度极限可达1nm。
实施例3、蛋白表达的控制
由上可知设计的dna传感器对dna序列的检测有着很高的特异性。除此之外,设计的dna传感器还可以作为蛋白表达控制的开关,
反应原理如图6所示,序列设计仍采用之前的序列设计方法,但是将a区的随机序列改为lacz基因序列。当dna聚合酶将变形后的probe探针修补为双链之后,将其放入到cell-free反应系统中进行反应,然后根据反应吸光度来检测反应的变化情况。
cell-free模板制备:通过pcr的方法在lacz基因序列中加入了探针序列和t7promoter序列。标准反应1×easytaq缓冲液,250μmdntps,0.2μm探针引物,0.2μm反向引物,1ngdna模板(pcr产物),2.5ueasytaqdna聚合酶,加水至50μl。热循环反应在94℃下开始5分钟并循环30次90℃30秒,在55℃下30秒,在72℃下30秒,然后在72℃下最终延伸7分钟。产物在2%琼脂糖凝胶上电泳,用溴化乙锭染色。接下来,使用plusdnaclean/extractionkit(genemark)获得dsdna模板。然后,使用不对称pcr制备ssdna。最后,使用zymocleangeldnarecoverykit(zymoresearch)来获得ssdna模板。将反应得到的120ngprobe-laczssdna在1×te缓冲液(tris-edta)中进行95℃变性3分钟并缓慢冷却至室温。加入1.5μmtrigger序列,温育37度,30min。在1×blue缓冲液中加入0.33u/μlklenowfragmentexo-和250μmdntps后,在37℃温育30分钟终止反应。纯化回收,将纯化回收后的产物放入到cell-free反应体系中,30℃反应3h。
1.单独加入单链probe-lacz;2.probe-lacz+trigger;3.probe-lacz+klenowfragmentexo-dna聚合酶;4.probe-lacz+trigger+klenowfragmentexo-dna聚合酶。在反应中不加入的物质以水代替。将这四个反应产物最后都加入到cell-free中进行反应,由上图可看出,当加入trigger后,吸光度增大,说明蛋白表达量增加,由此也说明设计的dna传感器对于控制蛋白表达起到了很好的效果。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>天津大学
<120>一种寡核苷酸组、一种核酸序列的扩增方法及目的蛋白的表达方法
<130>mp1826114
<160>4
<170>siposequencelisting1.0
<210>1
<211>63
<212>dna
<213>人工序列(artificialsequence)
<400>1
tgcttggtttcgaaatgtattgattcttgccttgttcgtttgaatcaatacatttcgtgt60
att63
<210>2
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>2
aaacgaacaaggcaagaatcttttt25
<210>3
<211>90
<212>dna
<213>人工序列(artificialsequence)
<400>3
ccctaaccctaaccctaaccctccttgactctgctcgaaatgtattgattcttgccttgt60
tcgtttgaatcaatacatttcgagtgtatt90
<210>4
<211>449
<212>dna
<213>人工序列(artificialsequence)
<400>4
ctatgcggcatcagagcagattgtactgagagtgcaccatatgcggtgtgaaataccgca60
cagatgcgtaaggagaaaataccgcatcaggcgccattcgccattcaggctgcgcaactg120
ttgggaagggcgatcggtgcgggcctcttcgctattacgccagctggcgaaagggggatg180
tgctgcaaggcgattaagttgggtaacgccagggttttcccagtcacgacgttgtaaaac240
gacggccagtgaattcgagctcggtacccggggatcctctagagtcgacctgcaggcatg300
caagcttggcgtaatcatggtcatatggtatatctccttcttaaagttaaacaaaattat360
tgctagccctatagtgagtcgtattagcgctgctcgaaatgtattgattcttgccttgtt420
cgtttgaatcaatacatttcgagtgtatt449
- 还没有人留言评论。精彩留言会获得点赞!
- 基于DNA酶嵌合引物的核酸筛查生物传感器的制造方法与工艺
- 一种高密度、高稳定性的核酸芯片及其制备方法与流程
- CYP2C9*3基因多态性快速检测的引物、分子信标、试剂盒及其检测方法与流程
- 黄牛ADNCR基因单核苷酸多态性的四引物扩增受阻突变体系PCR检测方法及其应用与制造工艺
- 一种原位杂交探针及其对大麦染色体组鉴定的方法与制造工艺
- 一种大麦亚端粒寡核苷酸探针及其应用的制造方法与工艺
- 一种核酸检测的复合物及其制备方法与核酸检测的方法与制造工艺
- 用于检测人类ROS1融合基因的核酸、试剂盒及方法与制造工艺
- 一种特异性结合GPC3的核酸适配体及其应用的制造方法与工艺
- 一种单链核酸分子、聚合酶活性测定方法及试剂盒与制造工艺