一种倍半萜环己烯酮类化合物及其制备方法和应用与流程
本发明涉及萜类化合物技术领域,尤其涉及一种倍半萜环己烯酮类化合物及其制备方法和应用。
背景技术:
萜类化合物(terpenes)-分子式为异戊二烯单位的倍数的烃类及其含氧衍生物,是具有高度多样性的一大类天然产物,包括单萜、倍半萜、二萜、倍半萜环己烯酮、三萜等等。许多萜类化合物已被发展为治疗癌症、细菌感染、疟疾和其他各种人类疾病的重要药物。因此,萜类化合物的合成就显得非常的重要。但由于生源合成途径仍未完全阐明、萜类化合物具有非模块化的结构以及没有普适的统一的合成策略等因素,萜类化合物的来源途径仍依赖于天然产物的提取。
倍半萜环己烯酮类化合物为极少数微生物的次生代谢产物,据文献报道,该类化合物具有抗炎、抗细胞毒、抗肿瘤以及抗菌等多种生物活性。因此,开发新型倍半萜环己烯酮类化合物,对解决目前抗生素滥用的现状具有重要意义。
技术实现要素:
本发明的目的在于提供一种倍半萜环己烯酮类化合物,该类化合物结构新颖,具有抗炎及抗肿瘤活性。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种倍半萜环己烯酮类化合物,具有式i或式ii所示结构:
其中,r为-oh或ch3coo-。
本发明提供了上述技术方案所述倍半萜环己烯酮类化合物的制备方法,包括以下步骤:
将扩展青霉接种于发酵培养基中进行发酵,得到扩展青霉发酵物;
将所述扩展青霉发酵物与醇混合,进行超声提取,得到扩展青霉宁粗提物;
将所述扩展青霉宁粗提物进行纯化,得到倍半萜环己烯酮类化合物。
优选的,所述发酵培养基的原料包括马铃薯或大米。
优选的,所述发酵的温度为20~30℃,时间为25~35d,所述发酵的方式为固体发酵。
优选的,所述扩展青霉发酵物的质量与醇的体积之比为(30~50)g:(50~100)ml。
优选的,所述醇为甲醇、乙醇、丙醇或异丙醇。
优选的,所述超声提取的时间为20~40min,功率为250~350w。
优选的,所述纯化的步骤包括先采用硅胶柱色谱进行洗脱,然后将所得洗脱液进行色谱纯化,得到倍半萜环己烯酮类化合物。
优选的,所述洗脱所用的洗脱剂为氯仿-甲醇和石油醚-丙酮,所述洗脱剂的流速为2~5ml/min。
本发明提供了上述技术方案所述倍半萜环己烯酮类化合物在制备抗炎和抗肿瘤药物中的应用。
本发明提供了一种倍半萜环己烯酮类化合物,该类化合物是具有新颖骨架结构的萜类化合物,丰富了倍半萜环己烯酮类化合物的多样性;
本发明提供的倍半萜环己烯酮类化合物具有抗炎和抗肿瘤活性,根据实施例可知,该化合物对no的生成均表现出了明显的抑制活性,且对5种肿瘤细胞表现出了明显的抑制活性;
本发明通过微生物发酵制备得到倍半萜环己烯酮类化合物,制备方法周期短、培养条件温和、副产物少、立体选择性强、成本低,易于实现工业化,不仅符合现代环保和低碳经济的需求,还能够为倍半萜环己烯酮类化合物的量产提供新途径;
本发明提供的倍半萜环己烯酮类化合物能够应用于制备抗炎及抗肿瘤药物,为开发抗炎药物提供了新的选择。
附图说明
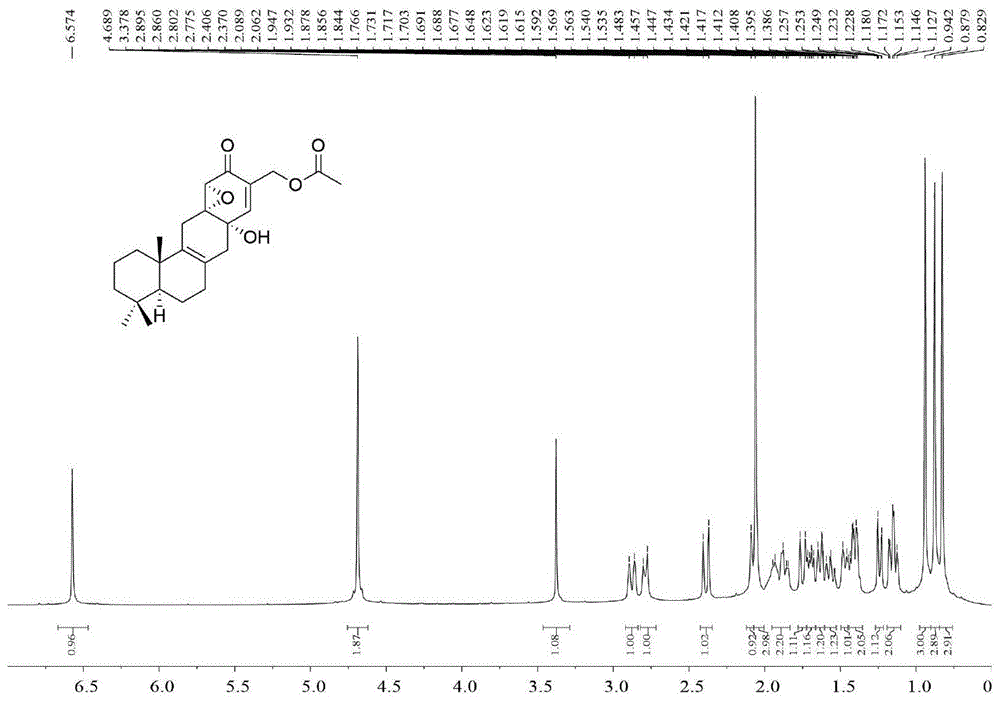
图1为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的1h-nmr谱;
图2为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的13c-nmr和dept谱;
图3为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的1h-1hcosy谱;
图4为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的hmbc谱;
图5为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的hsqc谱;
图6为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的noesy谱;
图7为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的hr-esi-ms谱;
图8为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲的x射线单晶结构;
图9为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的1h-nmr谱;
图10为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的13c-nmr和dept谱;
图11为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的1h-1hcosy谱;
图12为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的hmbc谱;
图13为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的hsqc谱;
图14为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的noesy谱;
图15为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁乙的hr-esi-ms谱;
图16为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁甲和乙的ecd实验图谱;
图17为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的1h-nmr谱;
图18为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的13c-nmr和dept谱;
图19为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的1h-1hcosy谱;
图20为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的hmbc谱;
图21为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的hsqc谱;
图22为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的noesy谱;
图23为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙的hr-esi-ms谱;
图24为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丙和丁的ecd实验图谱;
图25为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的1h-nmr谱;
图26为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的13c-nmr和dept谱;
图27为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的1h-1hcosy谱;
图28为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的hmbc谱;
图29为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的hsqc谱;
图30为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的noesy谱;
图31为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的hr-esi-ms谱;
图32为本发明实施例1制备的倍半萜环己烯酮类化合物扩展青霉宁丁的x射线单晶结构。
具体实施方式
本发明提供了一种倍半萜环己烯酮类化合物,具有式i或式ii所示结构:
其中,r为-oh或ch3coo-。
在本发明中,当式i所示结构的化合物中r为ch3coo-时,所述倍半萜环己烯酮类化合物记为扩展青霉宁甲;
当式i所示结构的化合物中r为-oh时,所述倍半萜环己烯酮类化合物记为扩展青霉宁乙;
当式i所示结构的化合物中r为ch3coo-时,所述倍半萜环己烯酮类化合物记为扩展青霉宁丙;
当式i所示结构的化合物中r为-oh时,所述倍半萜环己烯酮类化合物记为扩展青霉宁丁。
本发明提供了上述技术方案所述倍半萜环己烯酮类化合物的制备方法,包括以下步骤:
将扩展青霉接种于发酵培养基中进行发酵,得到扩展青霉发酵物;
将所述扩展青霉发酵物与醇混合,进行超声提取,得到扩展青霉宁粗提物;
将所述扩展青霉宁粗提物进行纯化,得到倍半萜环己烯酮类化合物。
本发明将扩展青霉接种于发酵培养基中进行发酵,得到扩展青霉发酵物。在本发明中,所述扩展青霉优选来源于黄草乌,更优选从黄草乌的根部分离得到,所述扩展青霉为黄草乌的内生真菌。本发明优选先将所述扩展青霉进行活化后,再进行发酵;所述活化的步骤优选为将所述扩展青霉接种到pda斜面培养基上,在25~30℃恒温培养5~7d后,得到活化的扩展青霉,将其置于5℃冰箱中储存备用。
在本发明中,所述发酵培养基的原料优选包括马铃薯或大米,更优选为马铃薯。本发明对所述发酵培养基没有特殊限定,能够使扩展青霉生长繁殖即可。在本发明的实施例中,具体是将马铃薯原料分成直径为1~3cm的块状(或将大米浸泡在水中)并置于组织培养瓶中(45~50g/瓶),然后将所述组织培养瓶加盖后在120~150℃下进行高温灭菌30~50min,冷却,得到发酵培养基。
在本发明中,所述接种的接种量优选为1~3%。本发明对所述接种的条件没有特殊的要求,选用本领域技术人员熟知的方式进行接种即可。在本发明的实施例中,具体是将活化的扩展青霉接种于发酵培养基中,加盖后进行恒温培养,得到扩展青霉发酵物。
在本发明中,所述发酵的方式优选为固体发酵,所述发酵的温度优选为20~30℃,更优选为26~28℃;时间优选为25~35d,更优选为28~30d。本发明采用固体发酵方式能够缩短发酵时间。
得到扩展青霉发酵物后,本发明将所述扩展青霉发酵物与醇混合,进行超声提取,得到扩展青霉宁粗提物。在本发明中,所述醇优选为甲醇、乙醇、丙醇或异丙醇;本发明以醇为溶剂将扩展青霉宁甲、乙、丙、丁从扩展青霉发酵物中提取出来。在本发明中,所述扩展青霉发酵物的质量与醇的体积之比优选为(30~50)g:(50~100)ml,更优选为(40~45)g:(70~80)ml,最优选为45g:80ml。本发明对所述混合的方式没有特殊的限定,选用本领域技术人员熟知的方式进行混合即可。
在本发明中,所述超声提取的时间优选为20~40min,更优选为30min,功率优选为250w~350w,更优选为300w。本发明优选在室温条件下进行所述超声提取。完成所述超声提取后,本发明优选将所得提取液进行过滤,然后去除所得滤液中的溶剂,得到扩展青霉宁粗提物。本发明优选采用减压蒸馏的方式去除滤液中的溶剂;在本发明的实施例中,具体是将滤液减压浓缩至无醇味,所述减压浓缩的压力优选为10~15kpa,更优选为12kpa;所述减压浓缩的温度优选为45~55℃,更优选为50℃。
得到扩展青霉宁粗提物后,本发明将所述扩展青霉宁粗提物进行纯化,得到倍半萜环己烯酮类化合物。
在本发明中,所述纯化的步骤优选包括先采用硅胶柱色谱进行洗脱,然后将所得洗脱液进行色谱纯化,得到倍半萜环己烯酮类化合物。本发明优选先以氯仿-甲醇溶液为洗脱剂进行洗脱,然后以石油醚-丙酮为洗脱剂进行洗脱。本发明所述氯仿-甲醇溶液中,氯仿与甲醇的体积比优选为100:0~20:1。在本发明的实施例中,具体是先用氯仿-甲醇溶液将扩展青霉宁粗提物溶解,将所得混合物与硅胶混合、去除溶剂、装柱,然后用氯仿为溶剂洗脱得到氯仿洗脱部分;以体积比30:1的石油醚-丙酮为洗脱剂对氯仿洗脱部分进行硅胶柱层析洗脱,得到含扩展青霉宁甲和乙的洗脱液,以体积比15:1的石油醚-丙酮为洗脱剂对氯仿洗脱部分进行硅胶柱层析洗脱,得到含扩展青霉宁丙和丁的洗脱液。在本发明中,用于溶解扩展青霉宁粗提物的氯仿-甲醇溶液优选为任意体积比的洗脱剂,本发明利用任意体积比的氯仿-甲醇溶液将扩展青霉宁粗提物溶解后吸附到硅胶上。在本发明中,所述硅胶与扩展青霉宁粗提物干重的质量比优选为0.8~1.5:1;所述硅胶的粒径优选为300~400目。在本发明中,所述去除溶剂的方法优选为减压蒸馏法;所述减压蒸馏的温度优选为48~55℃,更优选为50℃。在本发明中,进行所述洗脱时,洗脱剂的流速优选为2~5ml/min,更优选为3ml/min。
本发明优选采用凝胶柱色谱对所得洗脱液进行色谱纯化,具体是将所述含扩展青霉宁甲和乙的洗脱液、以及含扩展青霉宁丙和丁的洗脱液经葡聚糖凝胶柱进行纯化,得到扩展青霉宁甲、乙、丙和丁。在本发明中,进行所述纯化的溶剂优选为甲醇,所述甲醇的流速优选为0.4~0.6ml/min,更优选为0.5ml/min。完成所述纯化后,本发明优选将所得产物进行干燥,得到扩展青霉宁甲、乙、丙和丁。本发明对所述干燥的方式没有特殊的限定,选用本领域技术人员熟知的方式进行干燥即可。
本发明提供了上述技术方案所述倍半萜环己烯酮类化合物在制备抗炎和抗肿瘤药物中的应用。本发明提供的倍半萜环己烯酮类化合物具有显著的抗炎和抗肿瘤活性,能够应用于制备抗炎和抗肿瘤药物。在制备抗炎和抗肿瘤药物时,所述抗炎和抗肿瘤药物中优选包括质量百分数为1~99%的扩展青霉宁甲、乙、丙或丁,更优选为55~90%;所述抗炎和抗肿瘤药物中优选还可以包括药用辅料,本发明对所述药用辅料没有特殊的限定,选择本领域常规的制药辅料即可。本发明对所述抗炎和抗肿瘤药物的制备方法没有特殊的限定,采用本领域熟知的制备方法制成片剂、颗粒剂或注射剂等剂型均可。
下面结合实施例对本发明提供的倍半萜环己烯酮类化合物及其制备方法和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
将扩展青霉接种到pda斜面培养基上,28℃恒温培养5d后,得到活化的扩展青霉,置于5℃冰箱中储存备用;
取洗净的土豆,分成直径为1cm的土豆块并置于组织培养瓶中(45g/瓶),将组织培养瓶加盖后在120℃下进行高温灭菌30min,冷却,得到发酵培养基;
将所述活化的扩展青霉按照1%的接种量接种到所述发酵培养基中,加盖后在28℃下恒温培养30d,得到扩展青霉发酵物;
将50g所述扩展青霉发酵物与80ml甲醇混合,所得混合溶液在40khz条件下超声30min,过滤,取滤液进行减压浓缩至无醇味,得到27.0g扩展青霉宁粗提物;
用30ml氯仿-甲醇溶液(体积比为2:1)溶解27.0g扩展青霉宁粗提物,再与27.0g硅胶(300~400目)混合,减压浓缩去除溶剂,装柱;然后用氯仿为溶剂洗脱得到氯仿洗脱部分;以体积比30:1的石油醚-丙酮为洗脱剂对氯仿洗脱部分进行硅胶柱层析洗脱,得到含扩展青霉宁甲和乙的洗脱液,以体积比15:1的石油醚-丙酮为洗脱剂对氯仿洗脱部分进行硅胶柱层析洗脱,得到含扩展青霉宁丙和丁的洗脱液,以甲醇为溶剂,对所述扩展青霉宁甲、乙、丙和丁的洗脱液进行葡聚糖凝胶柱色谱纯化,干燥,得到倍半萜环己烯酮类化合物-扩展青霉宁甲、乙、丙和丁。
取实施例1制备得到的扩展青霉宁甲、乙、丙、丁,通过1d/2dnmr(一维核磁共振波谱和二维核磁共振波谱)以及hr-esi-ms(高分辨电喷雾电离质谱)进行结构鉴定。
由hsqc谱结合碳谱可以得到化合物扩展青霉宁甲、乙、丙、丁的h和与之相连接的c的化学位移δ归属,如表1。
表1扩展青霉宁甲、乙、丙、丁的13c(150mhz)和1h(600mhz)nmr数据,cdcl3为溶剂。
根据表1,化合物1的hr-esi-msm/z:[m+h]+的准分子离子峰为401.2323(c24h33o5[m+h]+计算值:401.2334)表明其分子式为c24h32o5,含有9个不饱和度。其中2个双键c-8=c-9和c-4′=c-5′,一个酮羰基(-c=o)和一个酯羰基(-coo-)共占有4个不饱和度,其余5个不饱和度表明化合物1具有五环骨架。化合物1的红外光谱在3444、1734和1712cm-1处的吸收峰表明化合物1中含有羟基和羰基。通过对化合物1的1h、13c、dept和hsqcnmr谱的分析可知化合物1中含有4个甲基[δh0.88(h3-13),0.83(h3-14),0.94(h3-15),2.06(h3-9′);δc33.2(c-13),21.7(c-14),19.4(c-15),20.9(c-9′)]、8个亚甲基[δh见表2;δc36.1(c-1),18.9(c-2),41.7(c-3),18.7(c-6),31.5(c-7),27.1(c-11),44.6(c-12),60.4(c-7′,oxygenated)]、3个次甲基[δh1.24(h-5),6.57(h-2′),3.38(h-5′);δc51.2(c-5),147.5(c-2′,olefinic),62.2(c-5′,oxygenated)]以及9个季碳[δc33.3(c-4),121.9(c-8,olefinic),136.4(c-9,olefinic),37.7(c-10),69.8(c-1′,oxygenated),129.0(c-3′,olefinic),193.5(c-4′,α,β-unsaturatedketone),63.3(c-6′,oxygenated),170.5(c-8′,ketone)]。
化合物1的平面结构通过核磁数据对比以及2dnmr实验确定。h2-1(δh1.15m,1.64m)/h2-2(δh1.42-1.48m,1.57m)/h2-3(δh1.37m,1.93m)以及h-5/h2-6(δh1.70m,1.42m)/h2-7(1.87-1.93m,2h)的1h-1hcosy相关结合h2-1/c-2,c-3,c-10和c-5;h2-2/c-3,c-4和c-10;h2-3/c-4和c-5;h-5/c-4,c-10,c-6和c-7;h2-6/c-4,c-10,c-7和c-8;h2-7/c-8,c-9;h3-13/c-3,c-4,c-5和c-14;h3-14/c-3,c-4,c-5和c-13以及h3-15/c-1,c-5,c-9和c-10的hmbc相关(图4)证实了一个双环倍半萜稀(ringsaandb)的存在。顶环氧菌素部分(环氧环己烯酮部分)(ringd)的存在则是根据以下hmbc相关确定:h-2′/c-12,c-3′,c-4′和c-6′;h-5′/c-3′,c-4′和c-6′;h2-7′/c-2′,c-3′,c-4′和c-8′以及h3-9′/c-8′。而h2-12/c-7,c-8,c-9,c-1′和c-6′以及h2-11/c-8,c-9,c-1′和c-6′的hmbc相关则表明倍半萜稀片段和顶环氧菌素部分是通过一个环己烯环(ringc)相连接。c-1′的化学位移值δc69.8表明其与一个羟基相连。因此,化合物1的平面结构被确定为由一个环己烯环连接的倍半萜环氧环己烯酮的聚合物,该化合物为一个由已知化合物环化衍生而得的新骨架化合物。化合物1的相对构型和绝对构型分别通过noesy实验(图6)和单晶x射线衍射实验确定。h3-14/h3-15,h3-15/hβ-11以及hβ-11/h-5′的noe相关表明h3-14,h3-15以及h-5′位于β位;而h3-13/h-5的noe相关则表明h3-13和h-5位于α位。通过单晶x射线衍射实验[cukα,flackparameter0.00(6)]确定了化合物1的绝对构型(图8)。因此,化合物1的结构得到完全确证,将其命名为扩展青霉宁甲。
化合物2的hr-esi-msm/z:[m+h]+的准分子离子峰为359.2215(c22h31o4[m+h]+计算值:395.2217)表明其分子式为c22h30o4,含有8个不饱和度。仔细对比化合物2和化合物1的1h和13cnmr谱,发现二者的一维谱几乎可以完全重叠,不同之处在于化合物2相较化合物1少了一个乙酰基的信号(δh2.06s,δc20.9,170.5),进一步分析其hmbc数据(图12),与化合物1相比较,化合物2中缺失了在化合物1中存在的h2-7′/c-8′以及h3-9′/c-8′的hmbc相关也表明化合物2中不存在乙酰基的结构片段。因此我们推测化合物2为化合物1的去乙酰化产物。由于化合物2与化合物1相比少了一个乙酰基的结构,化合物2的相对构型采用noesy试验确定(图14),h3-14(δh0.83s)/h3-15(δh0.94s),h3-15/hβ-11(δh2.88d)以及hβ-11/h-5′(δh3.38s)的noe相关表明h3-14,h3-15以及h-5′位于β位;而h3-13(δh0.88s)/h-5(δh1.24d)的noe相关则表明h3-13和h-5位于α位。由于2与化合物1有着相同的分子骨架,并且相对构型完全一致。为了进一步确定化合物2的绝对构型,对化合物1和2的实验ecd谱图进行比较,发现二者的ecd图谱几乎完全重叠(图16),因此,化合物2的绝对构型被确定为5s,10s,1′s,5′s,6′r,化合物2被命名为扩展青霉宁乙。
化合物3的hr-esi-msm/z:[m+na]+的准分子离子峰为423.2146(c24h32o5na[m+na]+计算值:423.2142)表明其分子式为c24h32o5,含有9个不饱和度。其中2个双键c-8=c-9和c-3′=c-2′,一个酮羰基(-c=o)和一个酯羰基(-coo-)共占有4个不饱和度,其余6个不饱和度表明化合物3具有六环骨架。化合物3的红外光谱在3439,1734和1718cm-1处的吸收峰表明化合物3中含有羟基和羰基。通过对其1h,13c,dept和hsqcnmr数据分析可知化合物3中含有4个甲基[δh2.08(h3-9′),0.94(h3-15),0.87(h3-13),0.81(h3-14);δc33.7(c-13),21.4(c-14),21.0(c-9′),20.9(c-15)],8个亚甲基[δh见表2;δc75.8(c-12,oxygenated),61.2(c-7′,oxygenated),42.3(c-3),39.5(c-1),33.3(c-7),26.4(c-11),19.1(c-6),18.2(c-2)],4个次甲基[δh6.84(h-2′),3.82(h-5′),1.84(h-9),0.97(h-5);δc135.5(c-2′,olefinic),58.4(c-9),57.3(c-5′),56.4(c-5)]以及8个季碳[δc194.4(c-4′,α,β-unsaturatedketone),170.5(c-8′),134.6(c-3′,olefinic),89.1(c-1′,oxygenated),69.3(c-6′),48.9(c-8),37.8(c-10),33.2(c-4)],以上波谱数据强烈表明化合物3为化合物1的类似物。
化合物3中h2-1(δh1.13m,1.58m)/h2-2(δh1.45m,1.66m)/h2-3(δh1.19m,1.43m)和h-5/h2-6(δh1.19m,1.75m)/h2-7(1.56m,2.31m)的1h-1hcosy相关以及h2-1/c-2,c-3,c-10和c-5;h2-2/c-2,c-4和c-10;h2-3/c-4,c-5和c-10;h-5/c-4,c-10,c-6和c-7;h2-6/c-4,c-10,c-7和c-8;h2-7/c-8,c-9;h3-11/c-3,c-4,c-5和c-12;h3-12/c-3,c-4,c-5和c-11以及h3-13/c-1,c-5,c-9和c-10的hmbc相关(图20)表明存在二环倍半萜稀的结构片段。h-2'/c-1',c-3',c-4'和c-7';h-5'/c-3',c-4'和c-6';h2-7'/c-3',c-4',c-5'和c-8'以及h3-9'/c-8'的hmbc相关表明化合物3中包含了顶环氧菌素的结构片段。h-7/c-1';h-2'/c-8以及h2-11(δh1.67overlapped,2.78t)/c-8,c-9,c-1',c-2'和c-6'的hmbc相关表明倍半萜部分和顶环氧菌素部分通过一个五元环连接。c-12和c-1'的化学位移值以及h2-12/c-7,c-8,c-9和c-1'的hmbc相关表明了一个四元氧环的存在。h3-14/h3-15;h3-15/hβ-11和h2-12(δh4.29d,4.82d);hβ-11/h-12(δh4.82d)和h-5'的noe相关表明这些基团位于β位,而h3-13/h-5以及h-9/h-5的noe相关表明这些基团位于α位。通过比较化合物3和化合物4的实验值ecd图谱(图24)结合化合物4的单晶结构(图32)也证明了化合物3的绝对构型,其结构如式i,化合物3被命名为扩展青霉宁丙。
化合物4的hr-esi-msm/z:[m+na]+的准分子离子峰为381.2038(c22h30o4na[m+na]+计算值:381.2036)表明其分子式为c22h30o4,含有8个不饱和度。对比化合物4和化合物3的1h和13cnmr谱可知化合物4相比化合物3少了一个乙酰基信号,此外,化合物3中的h2-7′/c-2′,c-3′,c-4′,和c-8′以及h3-9′/c-8′的hmbc相关(图28)的缺失也表明化合物4为化合物3的去乙酰化产物。同样的,乙酰基的缺失使得化合物4中c-3′的化学位移值向低场移动(δc134.6→138.8),因此,化合物4被确定为化合物3的去乙酰化衍生物。化合物4的相对构型通过noesy实验确定,h3-14(δh0.80s)/h3-15(δh0.94s);h3-15/hβ-11(δh2.77t)和h2-12(δh4.29d,4.82d);hβ-11/h-12(δh4.82d)和h-5′(δh3.79s)的noe相关表明这些基团位于β位,而h3-13(δh0.87s)/h-5(δh0.94d)以及h-5/h-9(δh1.84dd)的noe相关则表明这些基团位于α位。因此,化合物4的相对构型被确定为5s*,8r*,9r*,10s*,1′s*,5′s*,6′r*,与化合物3的相对构型一致。化合物4的绝对构型通过ecd实验和单晶x射线衍射确定,由于化合物4和化合物3有着相同的骨架和完全相同的相对构型,通过二者的实验ecd谱(图24)可以看出二者有着相同的绝对构型。此外,化合物4的单斜晶体在石油醚-丙酮(4:1,v/v)体系中得到,通过单晶x射线衍射得到了其结构[图32,flackparameter=-0.06(7)],由此也证实了其绝对构型为5s,8r,9r,10s,1′s,5′s,6′r。化合物4绝对构型的确定也验证了化合物3绝对构型的正确性。至此,化合物4的结构得以完全确证,化合物4被命名为扩展青霉宁丁。
综上所述,可以确定实施例1制备得到的化合物扩展青霉宁甲、乙、丙、丁的结构式为:
实施例2
将实施例1制备的化合物扩展青霉宁甲、乙、丙、丁进行抗炎及抗肿瘤活性筛选。
1、化合物扩展青霉宁甲、乙、丙、丁的抗炎活性:
一氧化氮具有广泛而重要的生物学调控功能,在炎症、肿瘤及心血管系统等均有重要作用。当免疫细胞遭受微生物内毒素、炎症介质等刺激时,会生成大量的诱导型一氧化氮合成酶,产生no进行免疫应答。因此,抑制no生成是化合物抗炎活性的直接指标。
(1)取对数生长期的小鼠单核巨噬细胞raw264.7(购自中科院上海细胞库),用1μg/mllps进行诱导刺激,同时加入实施例1制备得到的化合物扩展青霉宁甲、乙、丙、丁处理,化合物扩展青霉宁甲、乙、丙、丁的终浓度依次为0.195μm、0.391μm、0.781μm、1.563μm、3.125μm和6.250μm六个梯度,每个梯度设置三个平行样,25℃条件下培养。
设置不含药物组和l-nmma(总nos抑制剂)阳性药物组按照上述相同步骤进行处理,作为对照。
(2)细胞过夜(10h)培养后,在570nm处检测各组的no生成量;向过夜培养得到的培养液中加入mts进行细胞存活率检测,排除化合物对细胞的毒性影响。
no生成抑制率(%)=(不含药物组od570nm-药物组od570nm)/不含药物组od570nm×100%;
ic50(半数抑制浓度)按reed&muench法计算,得到化合物扩展青霉宁甲、乙、丙、丁的no抑制活性的ic50。
2、化合物扩展青霉宁甲、乙、丙、丁的抗肿瘤活性:
mts法检测细胞活性原理:mts为一种全新的mtt类似物,是一种黄颜色的染料。活细胞线粒体中琥珀酸脱氢酶能够代谢还原mts,生成可溶性的甲臜化合物,甲臜的含量可以用酶标仪在490nm处进行测定。在通常情况下,甲臜生成量与活细胞数成正比,因此,可根据光密度od值推测出活细胞的数目。
(1)接种细胞:用含10%胎牛血清的培养液(dmem或者rmpi1640)配成单个细胞悬液,以每孔3000~15000个细胞接种到96孔板,每孔体积100μl,贴壁细胞提前12~24小时接种培养。
(2)加入待测化合物溶液:化合物用dmso溶解,化合物以40μm、8μm、1.6μm、0.32μm、0.064μm浓度复筛,每孔终体积200μl,每种处理均设3个复孔。
(3)显色:37摄氏度培养48小时后,贴壁细胞弃孔内培养液,每孔加mts溶液20μl和培养液100μl;悬浮细胞弃100μl培养上清液,每孔加20μl的mts溶液;设3个空白复孔(mts溶液20μl和培养液100μl的混合液),继续孵育2~4小时,使反应充分进行后测定光吸收值。
(4)比色:选择492nm波长,多功能酶标仪(multiskanfc)读取各孔光吸收值,记录结果,数据处理后以浓度为横坐标,细胞存活率为纵坐标绘制细胞生长曲线,应用两点法(reedandmuench法)计算化合物的ic50值。
(5)阳性对照化合物:每次实验均设顺铂(ddp)和紫杉醇(taxol)两个阳性化合物,以浓度为横坐标,细胞存活率为纵坐标绘制细胞生长曲线,应用两点法(reedandmuench法)计算化合物的ic50值。
实验结果显示,在对化合物扩展青霉宁甲、乙、丙、丁的no抑制活性(阳性对照为:l-nmma)筛选试验中,化合物扩展青霉宁甲、乙、丙、丁对一氧化氮(no)的生成均表现出了明显的抑制活性(表2)。在对化合物扩展青霉宁甲、乙、丙、丁对体外五种肿瘤细胞的抑制活性(阳性对照为:顺铂,紫杉醇)筛选试验中,扩展青霉宁甲、乙、丙、丁均表现出了良好的抗肿瘤活性(表2)。因此,扩展青霉宁甲、乙、丙、丁具有作为先导化合物开发抗炎及抗肿瘤药物的研究价值。
表2扩展青霉宁甲、乙、丙、丁的no抑制活性及抗肿瘤活性筛选(ic50)
实施例3
将扩展青霉接种到pda斜面培养基上,28℃恒温培养5d后,得到活化的扩展青霉,置于5℃冰箱中储存备用;
取45g大米浸泡在40ml水中12h,取出大米并置于组织培养瓶中(50g/瓶),将组织培养瓶加盖后在120℃下进行高温灭菌30min,冷却,得到发酵培养基;
将所述活化的扩展青霉按照1%的接种量接种到所述发酵培养基中,加盖后在26℃下恒温培养25d,得到扩展青霉发酵物;
将45g所述扩展青霉发酵物与120ml甲醇混合,所得混合溶液在40khz条件下超声30min,过滤,取滤液进行减压浓缩至无醇味,得到27.0g扩展青霉宁甲、乙、丙、丁粗提物;
用30ml氯仿-甲醇溶液(体积比为2:1)溶解所述扩展青霉宁甲、乙、丙、丁粗提物,再与27.0g硅胶(300~400目)混合,减压浓缩去除溶剂,装柱;然后用氯仿为溶剂洗脱,得到氯仿洗脱部分;以体积比30:1的石油醚-丙酮为洗脱剂对氯仿洗脱部分进行硅胶柱层析洗脱,得到含扩展青霉宁甲和乙的洗脱液,以体积比15:1的石油醚-丙酮为洗脱剂对氯仿洗脱部分进行硅胶柱层析洗脱,得到含扩展青霉宁丙和丁的洗脱液,以甲醇为溶剂,对所述扩展青霉宁甲、乙、丙和丁的洗脱液进行葡聚糖凝胶柱色谱纯化,干燥,得到倍半萜环己烯酮类化合物-扩展青霉宁甲、乙、丙和丁。
由以上实施例可知,本发明提供了一种倍半萜环己烯酮类化合物,该类化合物是具有新颖骨架结构的萜类化合物,丰富了倍半萜环己烯酮类化合物的多样性;本发明提供的倍半萜环己烯酮类化合物具有抗炎和抗肿瘤活性,根据实施例可知,该化合物对no的生成均表现出了明显的抑制活性,且对5种肿瘤细胞表现出了明显的抑制活性;本发明通过微生物发酵制备得到倍半萜环己烯酮类化合物,制备方法周期短、培养条件温和、副产物少、立体选择性强、成本低,易于实现工业化,不仅符合现代环保和低碳经济的需求,还能够为倍半萜环己烯酮类化合物的量产提供新途径;本发明提供的倍半萜环己烯酮类化合物能够应用于制备抗炎及抗肿瘤药物,为开发抗炎药物提供了新的选择。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!