一种齐墩果烷型三萜皂苷及其提取分离方法和应用与流程
本发明属于医药
技术领域:
,具体涉及一种齐墩果烷型三萜皂苷及其提取分离方法和应用。
背景技术:
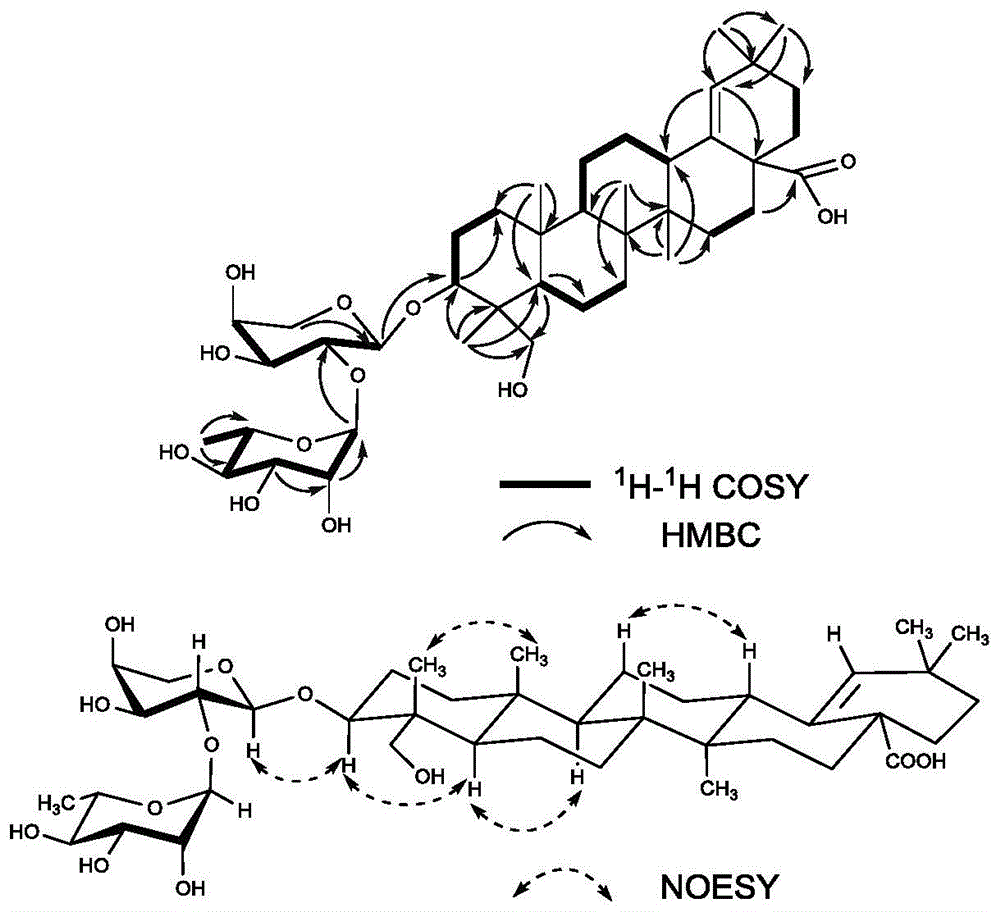
:腺毛黑种草(nigellaglandulifera)又名瘤果黑种草,为毛茛科(ranunculaceae)黑种草属(nigella)植物,主产地新疆,是新疆特有的植物资源,其种子是维吾尔族常用的药材,常用于胸闷气不足、乳肿、乳汁减少、耳鸣、热淋、石淋、闭经等症。近年来,对腺毛黑种草子化学成分和药理作用已有一些报道,主要有皂苷、黄酮、生物碱等生物活性成分,具有抗氧化、抗炎、免疫调节、抗肿瘤、降血脂等多种药理作用。维吾尔医报道除将黑种草子入药外,其根茎等亦可入药,但对其根茎化学成分的研究鲜有报道。对1类创新药物(单体化合物药物)研制的大量实践和报道证实:化学结构决定药理作用,新的化学结构预示着更强的生物活性、更低的毒副作用或更适于在临床使用。因此,迫切需要发现更多不同类别的新的化合物,以供进一步开发为高效低毒的抗癌新药。技术实现要素:本发明的目的在于提供一种齐墩果烷型三萜皂苷及其提取分离方法和应用。为了达到上述目的,本发明采用以下技术方案予以实现:本发明公开了一种齐墩果烷型三萜皂苷,所述齐墩果烷型三萜皂苷的分子式为c41h66o12,结构式如式ι:式ι所示的化合物的化学名称为3-o-α-l-吡喃鼠李糖基-(1→2)-α-l-吡喃阿拉伯糖基-3β,23-二羟基-18-双键-齐墩果酸,简称ngts。优选地,所述齐墩果烷型三萜皂苷是从腺毛黑种草中提取获得。本发明还公开了上述的齐墩果烷型三萜皂苷的提取方法,包括以下步骤:1)取腺毛黑种草的根茎,采用体积分数为70%的乙醇回流提取数次,合并提取液,回收溶剂,制得浸膏;2)将浸膏分散于水中,先用石油醚萃取数次,弃去石油醚层,再用水饱和正丁醇萃取数次,回收正丁醇层,制得总皂苷;3)将总皂苷上硅胶色谱柱,采用氯仿-甲醇-水的混合溶剂体系进行梯度洗脱,分离得到齐墩果烷型三萜皂苷粗品。优选地,步骤1)中,腺毛黑种草的根茎与体积分数为70%的乙醇的料液比为(10~30)kg:100l。优选地,步骤3)中,氯仿-甲醇-水的混合溶剂体系中氯仿、甲醇与水的体积比为20:1:0~6.5:3.5:1;洗脱时,每500ml洗脱液为一个收集流份。优选地,还包括对分离得到的齐墩果烷型三萜皂苷粗品进行纯化处理的操作,具体为:将齐墩果烷型三萜皂苷粗品先用sephadexlh-20凝胶柱分离纯化,再用纯甲醇洗脱,最后经高效液相色谱分离纯化,制得齐墩果烷型三萜皂苷纯品。进一步优选地,采用sephadexlh-20凝胶柱分离纯化,用纯甲醇洗脱,洗脱时每50ml为一个流份,合并第10~15个流份。进一步优选地,高效液相色谱分离纯化的色谱条件为:色谱柱选择ymc-packr&dods-a色谱柱20×250mm;以75%甲醇水溶液作为流动相,加0.1%三氟乙酸,流速6ml/min,25℃,在206nm下进行紫外检测。本发明还公开了上述的齐墩果烷型三萜皂苷在制备抗肿瘤药物中的应用。优选地,所述药物是指所述齐墩果烷型三萜皂苷与可药用载体制成的片剂、胶囊剂、注射剂、丸剂、散剂、颗粒剂、酊剂、膏剂或气雾剂。与现有技术相比,本发明具有以下有益效果:本发明从腺毛黑种草中分离出了一种结构新颖的齐墩果烷型三萜皂苷,经过光谱技术证实其是一种新的化合物,且是首次从腺毛黑种草中分离得到。如式ι所示的化合物ngts的苷元中双键结构位于18(19)位,在本属植物中比较罕见,在本属植物皂苷中首次发现。本发明公开了从腺毛黑种草中提取分离上述齐墩果烷型三萜皂苷的方法,该方法首先采用体积分数为70%乙醇为溶剂进行提取,合并提取液后回收溶剂得到浸膏,所用的提取溶剂毒性小,可回收循环利用,成本低,适合工业化生产;且提取过程对设备要求低,提取条件温和,操作较简便,提取效率高;其次,浸膏经过石油醚、水饱和正丁醇的萃取处理,得到总皂苷,总皂苷经过硅胶色谱柱,经过氯仿-甲醇-水的混合溶剂的梯度洗脱,获得齐墩果烷型三萜皂苷粗品。进一步地,对分离的齐墩果烷型三萜皂苷粗品先用sephadexlh-20凝胶柱分离纯化,再用纯甲醇洗脱,最后经高效液相色谱分离纯化,制得齐墩果烷型三萜皂苷纯品(即目标化合物ngts),两步分离纯化操作条件的筛选为成功分离、制备得到式i所示的化合物提供了有力支撑。本发明还首次公开了上述齐墩果烷型三萜皂苷具有抗癌作用,体外抗肿瘤试验发现该化合物对胃癌细胞和胶质瘤细胞有明显的抑制作用,表明其可以进一步作为新的抗肿瘤药物进行研究开发。附图说明图1为本发明的化合物ngts的1h-1hcosy、hmbc和noesy相关关系图谱。具体实施方式为了使本
技术领域:
的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。需要说明的是,本发明的说明书和权利要求书及上述附图中的术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的过程、方法不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法或其它步骤或单元。下面结合附图对本发明做进一步详细描述:1、化合物的提取和分离实施例1从腺毛黑种草中分离制备目标化合物(简称ngts),方法如下:原料为采自新疆乌鲁木齐市的腺毛黑种草,取腺毛黑种草的根茎部分20公斤,用铡刀切成小段后,加入100升70%的乙醇进行回流提取,每次2小时,共提取3次。合并提取液,减压回收乙醇,得浸膏2.5公斤。将浸膏分散于10升蒸馏水中,用等体积石油醚萃取3次,弃去石油醚层,然后用等体积水饱和正丁醇萃取3次,回收正丁醇层,得总皂苷225克。总皂苷用硅胶层析柱进行分离纯化,以体积比为20:1:0~6.5:3.5:1的氯仿-甲醇-水的混合溶剂梯度系统,每500ml洗脱液作为一个流份收集,并用硅胶薄层层析检测(展开剂为氯仿:甲醇:水=6:3:0.5,显色剂为10%的硫酸乙醇溶液,喷显色剂后于105℃加热显色),收集合并rf在0.5~0.7之间的流份,即为含化合物ngts的总皂苷混合物,蒸干溶剂得样品11.3克。样品用sephadexlh-20凝胶柱分离纯化,纯甲醇洗脱,每50ml为一个流份,薄层色谱分析,合并含ngts的第10至15个流份,蒸干溶液后得样品0.6克。最后经高效液相色谱分离纯化(hplc条件为:ymc-packr&dods-a色谱柱20×250mm,75%甲醇水溶液为流动相,加0.1%三氟乙酸,流速6ml/min,25℃,206nm紫外检测),得ngts纯品3.0毫克。实施例2从腺毛黑种草中分离制备目标化合物(简称ngts),方法如下:原料为采自新疆乌鲁木齐市的腺毛黑种草,取腺毛黑种草的根茎部分10公斤,用铡刀切成小段后,加入100升70%的乙醇进行回流提取,每次1小时,共提取3次。合并提取液,减压回收乙醇,得浸膏1.3公斤。将浸膏分散于5升蒸馏水中,用等体积石油醚萃取3次,弃去石油醚层,然后用等体积水饱和正丁醇萃取3次,回收正丁醇层,得总皂苷116克。总皂苷用硅胶层析柱进行分离纯化,以体积比为20:1:0~6.5:3.5:1的氯仿-甲醇-水的混合溶剂梯度系统,每500ml洗脱液作为一个流份收集,并用硅胶薄层层析检测(展开剂为氯仿:甲醇:水=6:3:0.5,显色剂为10%的硫酸乙醇溶液,喷显色剂后于105℃加热显色),收集合并rf在0.5~0.7之间的流份,即为含化合物ngts的总皂苷混合物,蒸干溶剂得样品6.1克。样品用sephadexlh-20凝胶柱分离纯化,纯甲醇洗脱,每50ml为一个流份,薄层色谱分析,合并含ngts的第10至15个流份,蒸干溶液后得样品0.31克。最后经高效液相色谱分离纯化(hplc条件为:ymc-packr&dods-a色谱柱20×250mm,75%甲醇水溶液为流动相,加0.1%三氟乙酸,流速6ml/min,25℃,206nm紫外检测),得ngts纯品1.6毫克。实施例3从腺毛黑种草中分离制备目标化合物(简称ngts),方法如下:原料为采自新疆乌鲁木齐市的腺毛黑种草,取腺毛黑种草的根茎部分30公斤,用铡刀切成小段后,加入100升70%的乙醇进行回流提取,每次2小时,共提取3次。合并提取液,减压回收乙醇,得浸膏3.6公斤。将浸膏分散于10升蒸馏水中,用等体积石油醚萃取3次,弃去石油醚层,然后用等体积水饱和正丁醇萃取3次,回收正丁醇层,得总皂苷268克。总皂苷用硅胶层析柱进行分离纯化,以体积比为20:1:0~6.5:3.5:1的氯仿-甲醇-水的混合溶剂梯度系统,每500ml洗脱液作为一个流份收集,并用硅胶薄层层析检测(展开剂为氯仿:甲醇:水=6:3:0.5,显色剂为10%的硫酸乙醇溶液,喷显色剂后于105℃加热显色),收集合并rf在0.5~0.7之间的流份,即为含化合物ngts的总皂苷混合物,蒸干溶剂得样品13.4克。样品用sephadexlh-20凝胶柱分离纯化,纯甲醇洗脱,每50ml为一个流份,薄层色谱分析,合并含ngts的第10至15个流份,蒸干溶液后得样品0.9克。最后经高效液相色谱分离纯化(hplc条件为:ymc-packr&dods-a色谱柱20×250mm,75%甲醇水溶液为流动相,加0.1%三氟乙酸,流速6ml/min,25℃,206nm紫外检测),得ngts纯品5.2毫克。2、化合物的结构鉴定实施例1式i化合物ngts,采用高分辨质谱和二维核磁共振谱等光谱技术对结构进行鉴定和解析:化合物ngts为白色无定型粉末(甲醇),liebermann-burchard和molish反应呈阳性,10%硫酸乙醇溶液显色反应呈紫色,表明其可能是三萜皂苷类化合物。esi-ms显示分子离子峰为m/z773[m+na]+(positivemode),hr-esi-ms给出准确分子量为m/z773.4430[m+na]+(计算值为773.4452),结合化合物的1dnmr数据得出化合物的分子式为c41h66o12。分析化合物ngts的1h-nmr谱,发现有6个单峰甲基氢,分别在δh0.68(3h,s,h-24)、δh0.82(3h,s,h-27)、δh0.93(3h,s,h-25)、δh0.97(3h,s,h-30)、δh0.98(3h,s,h-29)和δh1.02(3h,s,h-26),一个烯氢在δh5.12(s,h-19)和两个端基氢δh4.56(d,5.3,h-1’)和δh5.16(d,1.3,h-1”)。这是典型的齐敦果烷型三萜皂苷的氢谱数据。综合分析化合物的13c-nmr谱和hsqc谱,6个单峰甲基氢分别对应δc13.7、15.7、17.8、29.6、31.0和16.8,烯氢对应一个三取代双键δc133.6和139.0,两个端基氢分别对应端基碳δc104.5(c-1’)和102.1(c-1”)。此外,化合物的碳谱上还有一个化学位移δc82.4的次甲基碳信号,hsqc谱上与其相连的氢信号为δh3.62(h-3),表明其是糖链与苷元的连接位点;化学位移δc180.6的季碳信号,可能是羧酸的羰基碳信号。由此,推测化合物的苷元部分可能是齐墩果酸衍生物。在hmbc谱中,h-19与c-20、c-21、c-29、c-30、c-13和c-17相关,表明三取代双键位于c-18和c-19位;h-3与c-2、c-4、c-23、c-24和c-1’相关,表明糖链连接在化合物苷元的c-3位。通过化合物的1dnmr(1hnmr,13cnmr)和2dnmr(hsqc,hmbc,1h-1hcosy)谱数据对所有碳氢信号进行归属(表1)。表1.化合物ngts的1hnmr(800mhz,cd3od)和13cnmr(200mhz,cd3od)数据noesy谱中,h-3与h-1b和h-5相关,h-1b与h-9相关,表明h-3是α构型,h-23与h-25相关,表明h-23是β构型。通过酸水解和气相色谱分析化合物ngts中单糖的类型以及组成比例,结果显示l-鼠李糖(rha)和l-阿拉伯糖(ara)的比例为1:1。l-阿拉伯糖的端基氢耦合常数为j=5.3hz表明其绝对构型为α型;l-鼠李糖的端基氢耦合常数为j=1.3hz表明其绝对构型为α型。hmbc谱中,h-1”与c-2’相关,h-1’与苷元c-3相关,表明糖链连接在化合物苷元的c-3位,且rha与ara是1→2连接。结果参见图1。综上所述,确定化合物ngts的结构为3-o-α-l-吡喃鼠李糖基-(1→2)-α-l-吡喃阿拉伯糖基-3β,23-二羟基-18-双键-齐墩果酸,结构如下式:3、化合物体外抗肿瘤作用的研究对实施例1式i化合物ngts进行了体外抗肿瘤试验,试验所采用的细胞为人胃癌细胞sgc7901细胞和人胶质瘤u-251细胞(均购自中科院上海细胞研究所),采用常规的mtt法进行测试。具体方法如下:取在对数生长期生长状态良好的sgc7901细胞、u251细胞以及原代培养的人神经胶质细胞,调整细胞密度至103~104个/孔,每孔加入100μl。取细胞悬液接种于96孔板上,100μl/孔,置恒温co2培养箱中培养24小时后给药,待测化合物储备液用培养基稀释成浓度分别为0、2.5、5、10、20、40、80μmol/l,给药100μl/孔,每个浓度设5个复孔,继续培养72h后,每孔加5mg/mlmtt20μl培养4h。吸弃96孔板中孔内培养液,加入150μl/孔dmso,置摇床上低速振荡10min,用酶联免疫检测仪在波长为490nm处测定每孔的吸光值(od值),并计算受试药物对肿瘤细胞的抑制率(计算公式:细胞抑制率(inhibitoryrate)=(对照组od值-给药组od值)/(对照组od值-空白组od值))。实验结果ic50值是采用graphpadprism5.0软件计算。结果如下表2所示:表2对2种肿瘤细胞株sgc7901、u251以及神经胶质细胞的抑制作用(ic50值,μmol/l)化合物sgc7901u251神经胶质细胞ngts19.76±2.9815.43±2.56>100盐酸尼莫司汀0.98±0.08>100阿霉素0.62±0.05结果显示,化合物ngts对人胃癌细胞sgc7901细胞,人胶质瘤u-251细胞具有明显的抑制作用,其半数有效抑制浓度(ic50值)分别为19.76±2.98μmol/l和15.43±2.56μmol/l。实施例式i化合物ngts不影响原代培养的人神经胶质细胞生长(ic50值>100μmol/l),具体试验中发现,既使在实施例式i化合物ngts为100μmol/l浓度时,对神经胶质细胞的抑制率也只有8.2%(p>0.05),但阳性对照盐酸尼莫司汀在100μmol/l浓度时的抑制率可达28.3%(p<0.05),可见,实施例式i化合物ngts对正常神经胶质细胞基本无毒性。因此,将本发明的化合物ngts单独使用,或者与可药用载体一同制成片剂、胶囊剂、注射剂、丸剂、散剂、颗粒剂、酊剂、膏剂或气雾剂。以上内容仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明权利要求书的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1