一种调控PCV2病毒增殖的序列及其应用的制作方法
本发明属于分子生物学与生物医药领域,具体涉及一种调控pcv2病毒增殖的序列。
背景技术:
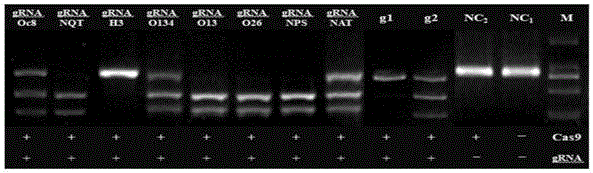
:crispr/cas9系统通过grna(tracrrna+crrna)与靶dnapam序列特异性识别,引发cas9核酸酶定点切割互补双链dna,在基因剪辑方面具有制备简单、剪辑位点特异性高及易于同时剪辑多个基因位点等优势,已被广泛应用于哺乳动物细胞基因剪辑。目前,crispr/cas9已成功应用于小鼠、斑马鱼和人类细胞甚至细菌的基因组精确编辑。crispr/cas9对病毒基因组的编辑相继也有报道,已成功应用crispr/cas9技术对双链dna病毒和单链rna病毒进行了有效编辑。目前尚未有应用crispr/cas9技术对单链dna病毒进行编辑的报道。猪圆环病毒(porcinecircovirus,pcv)是目前已知的侵染哺乳动物的最小病毒,属于圆环病毒科圆环病毒属,是一种无囊膜的单股环状dna病毒。猪圆环病毒1型(pcv1)和猪圆环病毒2型(pcv2)是猪圆环病毒的两个主要基因型。pcv1是从猪肾上皮细胞pk-15中分离出来的非致病性病毒粒子,pcv2发现于1998年,是造成猪圆环病毒相关病(porcinecircovirus-associateddisease,pcvad)的主要病原体。pcvad起初被描述为断奶仔猪多系统衰竭综合征(postweaningmultisystemicwastingsyndrome,pmws)。随着越来越多与pcv2感染相关的临床症状(诸如繁殖障碍、肠道疾病及呼吸症状)的出现,才将这种疾病改命名为猪圆环病毒相关病(美国)和猪圆环病毒病(欧洲)。作为一种大约只有1.7kb大小的单链环状dna病毒,pcv2的复制很大程度上依赖于宿主细胞的运转。因此,在病毒复制的过程中,pcv2通过与多种宿主细胞因子的相互作用来调节宿主免疫功能,从而导致细胞因子失衡,免疫抑制乃至疾病。pcv2基因组dna有11个潜在开放阅读框。间隔区ir含有与复制起点相关的茎环结构。由于pcv2极小的基因组限制了其编码能力,因此pcv2的生命周期很大程度上取决于其自身的复制能力。形态学上,pcvs粒子直径大约为15-20nm,无囊膜,二十面体对称的衣壳蛋白包裹整个基因组。pcvs的病毒粒子是包含60个衣壳蛋白拷贝的结构。pcv1和pcv2基因组内含11个潜在orfs。在这些orfs里,orf1和orf2是两个主要的orfs,并沿着相反的方向编码,这样的编码方式就衍生出一个双义基因组结构和2个基因间区域(ir)。其中,orf1和orf2基因的3’末端之间的ir是比较短的基因间区域,而它们的5’端之间的ir是较长的基因间区域,该区域内包含着pcv2复制的起点位置。pcv2复制起始于一个假定的茎环结构(stem-loop,sl),并以滚环复制模式进行复制。orf1和orf2之间间隔区内假定的茎环结构(stem-loop)和3个六核苷酸重复序列(h1,h2,h3:cggcag)与dna复制起始有关。茎环结构为反向重复序列,环区包含了10个核苷酸,含有保守的八聚体(oc8:a1x2t3a4x5t6↓a7c8;箭头代表dna复制时的切开位点)。orf1编码复制蛋白rep,orf2编码衣壳蛋白cap,orf3编码非结构蛋白由。pcv2的rep蛋白大小为35.8kda(314aa),由orf1全长编码。含有3个糖基化位点,23-25aa(nps);256-258aa(nqt)和286-288aa(nat)。为了间接控制管理pcvad,pcv2的复制性研究至关重要。降低pcv2病毒增殖的序列可以用于防治管理pcvad的传播或者发生、发展。技术实现要素:针对缺乏猪圆环病毒2型调控序列的问题,本发明提供一种调控pcv2病毒增殖的序列,可以有效调控pcv2的增殖。为实现上述目的,本发明以crispr/cas9系统原理及其grna的设计原理为基础,以px458为表达载体,获得pcv2cas9/grnas重组质粒,并对阳性cas9/grnas克隆进行了测序鉴定。本发明采用如下技术方案。一种调控猪圆环病毒2型(pcv2)病毒复制的grna序列,其碱基序列如seqidno:1-7任一所示。所述调控为抑制调控。所述grna序列,seqidno:1靶向pcv2复制起点的八聚体结构,碱基序列简称为grna-oc8。seqidno:2靶向orf1,orf3和orf4的重叠区,碱基序列简称为grna-o134。seqidno:3靶向orf1和orf3的重叠区,碱基序列简称为grna-o13。seqidno:4靶向orf2和orf6的重叠区,碱基序列简称为grna-o26。seqidno:5靶向orf1编码23-25aa的糖基化位点nps,碱基序列简称为grna-nps。seqidno:6靶向orf1编码256-258aa的糖基化位点nqt,碱基序列为grna-nqt。seqidno.7靶向orf1编码286-288aa的糖基化位点nat,碱基序列简称为grna-nat。上述grna序列对应的pam序列依次为:cgg、tgg、tgg、ggg、agg、agg、cgg。一种含上述grna的pcv2cas9/grnas重组质粒。一种上述pcv2cas9/grna重组质粒转染的细胞系。所述转染过程为:重组cas9/grnas转染细胞的同时,用等量的pcv2-sd病毒进行感染。一种上述grna、重组质粒和转染细胞系在制备治疗和预防猪圆环病毒2型病毒引起病症药物中的用途。本发明具有以下优点:本发明提供的调控猪圆环病毒2型(pcv2)病毒复制的grnas序列grna-oc8,grna-o13,grna-o134,grna-o26,grna-nps,grna-nqt和grna-nat,分别靶向pcv2复制起点的八聚体结构、orf1和orf3的重叠区、orf1,orf3和orf4的重叠区、orf2和orf6的重叠区、orf1编码23-25aa的糖基化位点nps、orf1编码256-258aa的糖基化位点nqt及orf1编码286-288aa的糖基化位点nat。通过构建的重组cas9/grnas,能对pcv2基因组进行有效编辑并抑制pcv2在pk-15细胞内的增殖复制。附图说明图1为grnas序列在pcv2基因组中的靶位点;图2为grnas序列特异性比对;图3为体外酶切电泳结果。m是dl-2000dnamarker;nc1是未经cas9酶切的转录产物;nc2是经cas9酶切的未转录产物;g1是经cas9酶切的标准grna1;g2是经cas9酶切的标准grna2;grna-oc8、grna-h3、grna-o13、grna-o134、grna-o26、grna-nps、grna-nqt和grna-nat是经cas9酶切的grnas;图4为不同重组cas9/grnas感染pk-15细胞48h后肉眼可见的细胞增殖情况;图5为重组cas9/grnas的蛋白表达结果。m是蛋白marker;oc8是重组pcv2cas9/grna-oc8;o134是重组pcv2cas9/grna-o134;o13是重组pcv2cas9/grna-o13;h3是重组pcv2cas9/grna-h3;o26是重组pcv2cas9/grna-o26;nat是重组pcv2cas9/grna-nat;nqt是重组pcv2cas9/grna-nqt;nps是重组pcv2cas9/grna-nps;nc是pcv2px458共转染作阴性对照;图6为重组cas9/grnas细胞毒性增殖实验结果。具体实施方式下面结合实施例和附图对本发明做进一步说明,但本发明不受下述实施例的限制。实施例1靶向pcv2基因组的grnas设计1靶向pcv2基因组的grnas设计根据ncbi中genbank上的pcv2sd株基因序列(genbankaccessionnumberjq653449)及pcv2全基因组结构,设计与pcv2复制相关的8个grna靶向片段:grna-oc8,grna-h3,grna-o13,grna-o134,grna-o26,grna-nps,grna-nqt和grna-nat,分别靶向pcv2复制起点的八聚体结构、茎环结构侧臂的六聚体、orf1和orf3的重叠区、orf1,orf3和orf4的重叠区、orf2和orf6的重叠区、orf1编码23-25aa糖基化位点nps的片段、orf1编码256-258aa糖基化位点nqt的片段及orf1编码286-288aa糖基化位点nat的片段,如图1所示。使用张峰等提供的crispr/cas9grna设计网站(http://crispr.mit.edu/)进行grnas的设计。将获得的grna进行blast比对筛选,通过筛选获得的grnas应具有以下特征:1)pcv2不同基因型的基因组共有序列中,有相同的碱基构成,确保grna具有靶向pcv2各基因型保守序列的能力;2)3’端为“ngg”,确保精准识别pam序列,进而确保pcv2序列能被cas9内切酶识别和切割。筛选后的grnas序列如表1。表1grnas序列信息namegrnasequence(5'-3')pamnucleotidepositiongrna-oc8ttaccagcgcacttcggcagcgg1766-21grna-h3ttgctgctgaggtgctgccgatg30-53grna-o134tgtcagaaatttccgcgggctgg479-502grna-o13ggcaacttactgatagaatgtgg351-374grna-o26gacagtataaccgatggtgcggg1430-1407grna-npsgcggaccccaaccacataaaagg76-99grna-nqtagaatactgcgggccaaaaaagg750-773grna-natcagctgtagaagctctctatcgg855-878通过ncbiblast序列比对,结果见图2。由图2可知,grna-oc8,grna-h3,grna-o13,grna-o134,grna-o26,grna-nps,grna-nqt和grna-nat对pcv2均具有较好的特异性。2体外转录grnas2.1grnaspcr产物的获得根据grnas的靶向位点(加粗部分,不含pam序列)合成不同引物f和引物r,序列如表2所示。表2grnas转录引物序列信息用grna-oc8-f和r、grna-h3-f和r、grna-o134-f和r、grna-o13-f和r、grna-o26-f和r、grna-nps-f和r、grna-nqt-f和r、grna-nat-f和r八对引物对,以标准grna片段为模板做pcr,同时根据spcas9-grna靶点效率检测试剂盒(北京唯尚立德生物科技有限公司,产品编号:no.vk007)准备标准grna1(g1)和标准grna2(g2)的pcr产物。通过cas9/grna体外酶切靶点dna片段,进行grna靶点活性的评估。pcr反应体系如下:组分用量grnas10nggrnas-f(10μm)1.5μlr(10μm)1.5μl2×pfumix25μlddh2o补至50μl反应程序为:预变性95℃3min,变性94℃30s,退火58℃30s,延伸72℃30s,共35个循环;最后72℃延伸10min,之后16℃10min。pcr产物进行凝胶电泳,回收120bp左右的目的条带,作为后续体外转录的dna模板。将grnas靶点(含pam序列)通过搭桥pcr的方式构建上述dna模板中。2.2cas9/grna体外酶切反应酶切前序列为:nnnnnnnnn(270bp)mmmmmmmmmmm(grna靶位点位置,含pam序列)nnnnnnnnnnn(450bp),酶切之前片段大小为740bp,酶切之后目的条带大小应分别为:280bp+460bp左右。反应体系如下:组分用量cas9酶1u10×cas9buffer2μlgrna(体外转录)50ng(grnas)ddh2o补至20μl酶切的dsdna50ng充分混合后,37℃反应0.5h,65℃煮5min后产物进行琼脂糖凝胶电泳,检测分析酶切结果,体外酶切电泳结果如图3,与标准grna1和标准grna2对比计算活性。根据酶切条带的灰度换算grna活性,如表3。标准grna1(g1)和标准grna2(g2)经过ssaluciferase试剂盒检测活性,其ssa活性分别为:标准grna1(g1)=3,标准grna2(g2)=10。因此,若样品grna的酶切效率<标准grna1,表明样品grna靶点活性比较差;若40%-50%≥样品grna的酶切效率≥标准grna1,表明样品grna靶点活性合格;若标准grna2≥样品grna的酶切效率≥40%-50%,表明样品grna靶点活性良好;若样品grna的酶切效率≥标准grna2(g2),表明样品grna靶点活性非常高。表3grnas体外活性检测结果名称靶序列体外酶切活性与标准品对照grna-oc8ttaccagcgcacttcggcagcgg70%等于grna2grna-h3ttgctgctgaggtgctgccgagg1%小于grna1grna-o134tgtcagaaatttccgcgggctgg73%大于grna2grna-o13ggcaacttactgatagaatgtgg98%大于grna2grna-o26gacagtataaccgatggtgcggg98%大于grna2grna-npsgcggaccccaaccacataaaagg95%大于grna2grna-nqtagaatactgcgggccaaaaaagg94%大于grna2grna-natcagctgtagaagctctctatcgg70%等于grna2由表3可见,除grna-h3外的所有grnas都有良好的活性。实施例2适用于编辑pcv2基因组的crispr/cas9体系的建立1pcv2cas9/grnas重组质粒构建1.1合成grnas二聚体根据实施例1设计好的grnas合成其反向互补引物对,如表4所示:表4crisprgrnas互补引物对序列信息名称正义链sense(5’-3’)反义链anti(5’-3’)grna-oc8ttaccagcgcacttcggcagctgccgaagtgcgctggtaagrna-h3ttgctgctgaggtgctgccgcggcagcacctcagcagcaagrna-o134tgtcagaaatttccgcgggcgcccgcggaaatttctgacagrna-o13ggcaacttactgatagaatgcattctatcagtaagttgccgrna-o26gacagtataaccgatggtgcgcaccatcggttatactgtcgrna-npsgcggaccccaaccacataaatttatgtggttggggtccgcgrna-nqtagaatactgcgggccaaaaatttttggcccgcagtattctgrna-natcagctgtagaagctctctatatagagagcttctacagctg将合成的grnas引物对稀释成10μm,按如下比例混合:组分用量grna-sense1μlgrna-anti1μlt4pnk5μlddh2o3μl混匀后,按如下程序处理:将pcr管放在95℃水中,自然冷却至室温。1.2重组cas9/grnas的构建将本实施例中步骤1.1所得产物grnas二聚体加入到如下体系中:组分用量px458质粒1μlgrnas二聚体2μlddh2o7μl充分混匀后,25℃静置5min。1.3转化将本实施例中步骤1.2的产物转化到stbl3感受态细胞中。1.4质粒的提取在培养良好的lb平板上随机挑取5个单菌落,分别放入含有5ml带有amp抗性的lb液体培养基中,37℃200rpm摇床上振荡培养16h,然后提取质粒,置于-20℃保存。1.5阳性cas9/grnas克隆的鉴定将本实施例中步骤1.4所得重组质粒dna进行测序鉴定,测序引物:tgagcgtcgatttttgtgatgctcgtcag。2.cas9/grnas重组载体功能验证2.1转染细胞的准备取装有pk-15细胞的冻存管,复苏后,在含有10%胎牛血清的dmem细胞培养液培养,用胰酶处理后吹打成单个细胞,将细胞培养液转移至6孔细胞培养板中,6孔板每孔加入2ml,其密度为0.25×106~1×106;置co2培养箱中,37℃培养,备用。2.2转染及转染后细胞生长情况转染操作按照lipofectaminetm3000试剂盒说明书进行。采用两种方式:(1)重组cas9/grnas转染细胞的同时,用等量的pcv2-sd病毒进行感染,对照组加入等量的空px458质粒,把细胞板放入co2培养箱中继续培养至48h;(2)重组cas9/grnas转染细胞24h后,用等量的pcv2-sd病毒进行感染,对照组加入等量的空px458质粒,把细胞板放入co2培养箱中继续培养至48h。用胰酶分别消化每孔细胞,制备细胞悬液,进行细胞计数,细胞悬液细胞数/ml=4个大格细胞总数/4×10000,所得实验数据经spss软件进行差异显著性分析。对两种不同的病毒感染方式进行了比较后发现,在重组cas9/grnas转染细胞的同时,方式(1):用等量的pcv2-sd病毒进行感染的比方式(2):在重组cas9/grnas转染细胞24h后,用等量的pcv2-sd病毒进行感染更容易产生肉眼可见的细胞生长上的变化。重组质粒和病毒同时转染48h后同一视野下的细胞生长情况,见图4。pcv2可以感染pk-15细胞,虽然不能形成细胞病变,但能影响pk-15细胞的增殖速度。由图4可见,同一批pk-15细胞在相同的生长条件下,对照组用空px458和pcv2-sd感染的细胞相比自然生长的纯pk-15细胞生长缓慢;感染pcv2-sd和重组cas9/grna-oc8、cas9/grna-o134、cas9/grna-o13、cas9/grna-o26、cas9/grna-nps和cas9/grna-nqt的细胞明显比对照组增殖快;重组cas9/grna-nat和pcv2-sd感染的细胞生长相比其他重组cas9/grnas缓慢,但比对照组生长较快;重组cas9/grna-h3和pcv2-sd同时感染的细胞增殖和对照组相差不大。方法(1)获得的重组cas9/grnas与pcv2共转染pk-15细胞48h后,对细胞数量的影响见表5:表5不同重组pcv2cas9/grnas对pk-15增殖数量的影响组别细胞数量oc835.742±1.855bh324.188±0.964ao13433.232±0.753bo1333.745±0.677ao2631.223±0.875bnps33.344±1.853bnqt33.875±0.289bnat28.098±1.584c对照23.542±0.875a注:标有相同字母的表示差异不显著(p>0.05);标有不同字母的表示差异显著(p<0.05)。细胞计数统计学分析显示,不同的重组pcv2cas9/grnas转染pk-15细胞48h后,重组pcv2cas9/grna-h3的细胞数量与对照组转染pcv2px458空质粒差异不显著;重组pcv2cas9/grna-oc8、pcv2cas9/grna-o134、pcv2cas9/grna-o13、pcv2cas9/grna-o26、pcv2cas9/grna-nps和pcv2cas9/grna-nqt的细胞数量与对照组差异显著;重组pcv2cas9/grna-nat的细胞数量与对照组差异显著并与其余重组pcv2cas9/grnas差异显著。2.3蛋白表达检测转染第48h时,取出细胞培养板,弃去营养液,经裂解、离心,取上清液至无菌的ep管中,加入sds-page上样缓冲液,进行sds-page电泳。获得的胶小心的取出,切除上层浓缩胶,切除边缘多于部分,依次经转印、封闭、洗膜、一抗孵育、洗膜、二抗孵育、洗膜、显影等步骤,用ecl超敏化学发光显色试剂盒进行显色,在chemidocmp全能型凝胶成像系统上曝光观察。蛋白免疫印迹结果见图5。由图5可见,重组pcv2cas9/grna-h3和对照组表达的rep蛋白量相差不大;重组pcv2cas9/grna-nat能表达一定量的蛋白,但蛋白表达量比对照组少;重组pcv2cas9/grna-nps、pcv2cas9/grna-nqt和pcv2cas9/grna-o26也显示出了蛋白的表达,但是表达量远远低于对照组,甚至低于重组pcv2cas9/grna-nat的蛋白表达量;重组pcv2cas9/grna-o134和pcv2cas9/grna-o13的蛋白表达量微乎及微;重组pcv2cas9/grna-oc8几乎无法表达rep蛋白。2.4pcv2cas9/grnascck-8毒力试验采用cck-8法进行细胞增殖和毒性分析:96孔板转染48h后,向每孔加入10μlcck-8溶液(避免在孔中生成气泡),将培养板在培养箱内孵育1-3h,分别在加入后0h、1h、2h时用酶标仪测定在450nm处的吸光度。加入cck-8后0h时测定吸光度作为参照。结果见图6,由图6可知,除重组pcv2cas9/grna-oc8和pcv2cas9/grna-h3在0h吸光度比对照组低之外,其他pcv2cas9/grnas的吸光度均高于对照组;所有重组pcv2cas9/grnas在1h时的吸光度均高于对照组,pcv2cas9/grna-h3的吸光度与对照组相近;除重组pcv2cas9/grna-h3在2h吸光度比对照组略低之外,其他pcv2cas9/grnas的吸光度均高于对照组。大量的研究表明,crispr/cas9技术存在一定程度的脱靶效应。影响基因编辑效率和特异性的因素多种多样。目前科学家们对crispr/cas9系统介导的基因组编辑效率和脱靶效应的研究尚不够深入,观点尚不统一。在各种grna设计与脱靶预测软件中,grna活性和特异性的评估标准也不一致。虽然目前存在多种crispr序列设计工具,但许多软件程序完全依赖于计算分析,没有真实可见的验证。试验筛选鉴定有效的grna是影响crispr/cas9编辑效率的关键因素。在本实验中应用在线网站分别对这8个不同的靶位点各设计了若干grnas,随后对从这些grnas中再挑选评分高的8个grnas,通过对这些grnas体外验证试验,结果证明,除了针对间隔区茎环结构侧臂上的六聚体合成的grna-h3体外活性低之外,其他7个grnas均具有较高的剪切活性。这也说明了试验筛选鉴定是确定grna有效性的唯一手段。序列表<110>山东省农业科学院畜牧兽医研究所<120>一种调控pcv2病毒增殖的序列及其应用<130>20190423<160>7<170>patentinversion3.5<210>1<211>20<212>dna<213>porcinecircovirus<400>1ttaccagcgcacttcggcag20<210>2<211>20<212>dna<213>porcinecircovirus<400>2tgtcagaaatttccgcgggc20<210>3<211>20<212>dna<213>porcinecircovirus<400>3ggcaacttactgatagaatg20<210>4<211>20<212>dna<213>porcinecircovirus<400>4gacagtataaccgatggtgc20<210>5<211>20<212>dna<213>porcinecircovirus<400>5gcggaccccaaccacataaa20<210>6<211>20<212>dna<213>porcinecircovirus<400>6agaatactgcgggccaaaaa20<210>7<211>20<212>dna<213>porcinecircovirus<400>7cagctgtagaagctctctat20当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1