一种噻吩并噻吩类的电致变色聚合物及其制备方法与流程
本发明涉及高分子材料领域,尤其涉及一种噻吩并噻吩类的电致变色聚合物及其制备方法。
背景技术:
1961年platt首次提出电致变色的定义,指材料在外界电信号驱动下引发可逆的氧化还原反应,其自身光学性质发生可逆变化的现象。几十年来随着对电致变色材料与器件的不断探究,该技术已经实现部分的商业化应用,应用领域众多,包括电纸书、智能标签、汽车后视镜、智能窗户等。
关于电致变色材料的种类分为无机和有机电致变色材料两大类。相对无机和有机小分子电致变色材料来说,聚合物电致变色材料具有成本低、对比度高、导电性高、颜色易调控、可实现柔性加工等优点,受到了广泛的关注与研究。目前关于聚合物材料的研究重点在于材料颜色调控这方面,因此许多不同颜色转变的新型聚合物电致变色材料已经得到研发,但他们在实际商业化应用中受离子扩散速率慢、响应时间久、稳定性差等所限制。为了提高材料离子导电性和响应速度,已报导的方法主要是将材料嫁接到碳纳米管上或制备成纳米结构,以增加材料与电解质的接触面积,然而这些方法对形貌调控的要求较高,且不适用于多种聚合物材料。
因此,发展离子扩散速率快、响应时间短、长循环稳定性的有机电致变色材料对实现多种材料的商业化发展等具有极其重要的意义。开发高性能的有机电致变色材料成为亟需解决的问题。
因此,现有技术还有待于改进和发展。
技术实现要素:
鉴于上述现有技术的不足,本发明的目的在于提供一种噻吩并噻吩类的电致变色聚合物及其制备方法,旨在解决由于现有电致变色材料离子扩散速率慢、响应时间长以及循环稳定性差的问题。
本发明的技术方案如下:
一种噻吩并噻吩类的电致变色聚合物,其中,其分子结构通式为:
一种噻吩并噻吩类的电致变色聚合物的制备方法,其中,包括步骤:
将噻吩并[3,2-b]噻吩、冰醋酸、氯仿和液溴混合搅拌,加热回流处理后,制得四溴噻吩并[3,2-b]噻吩化合物;
将锌单质与所述四溴噻吩并[3,2-b]噻吩化合物混合,再加入冰醋酸在室温条件下搅拌过夜,制得3,6-二溴噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将所述3,6-二溴噻吩并[3,2-b]噻吩化合物、溴化亚铜、碘化钾(41.47mg,0.25mmol)加入到甲醇钠溶液中,加热回流处理后,制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将所述3,6-二甲氧基噻吩并[3,2-b]噻吩化合物、对甲苯磺酸钠以及三乙二醇单乙醚加入甲苯溶剂中,加热回流处理后,制得分子结构式为
在氮气保护环境下,将化合物
有益效果:本发明提供了一种噻吩并噻吩类的电致变色聚合物,其分子结构通式为:
附图说明
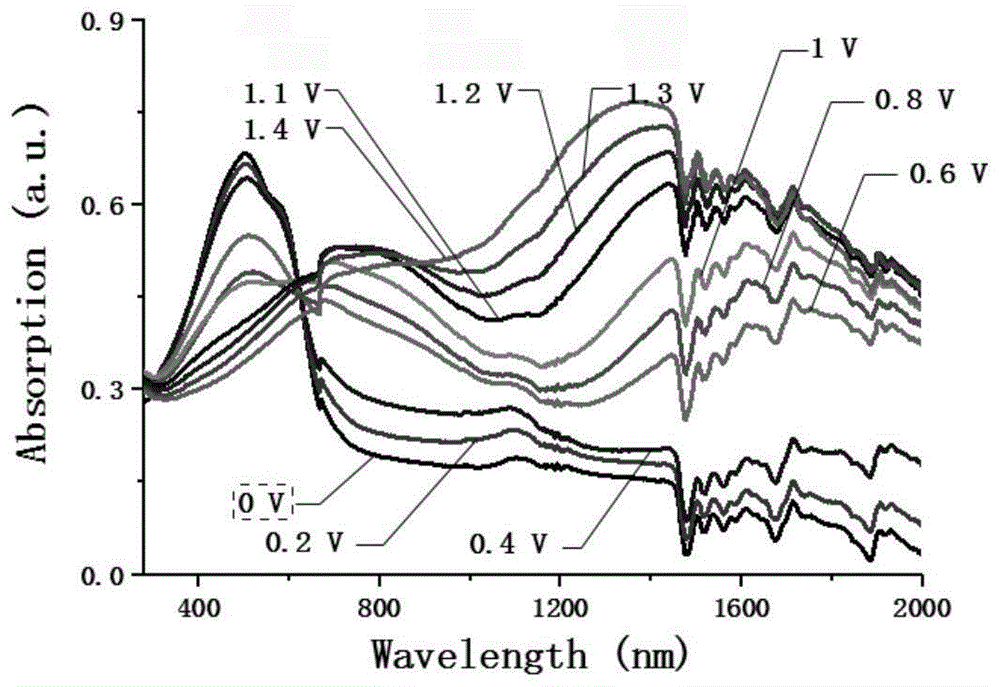
图1为本发明实施例1合成得到的新型噻吩并噻吩类的电致变色聚合物的光谱电化学图谱。
图2为本发明实施例1合成得到的新型噻吩并噻吩类的电致变色聚合物的方波函数图。
图3为本发明实施例5合成得到的新型噻吩并噻吩类的电致变色聚合物的光谱电化学图谱。
图4为本发明实施例5合成得到的新型噻吩并噻吩类的电致变色聚合物在700nm波长条件下的方波函数图。
图5为本发明实施例5合成得到的新型噻吩并噻吩类的电致变色聚合物在400nm波长条件下的方波函数图。
图6为本发明实施例7合成得到的新型噻吩并噻吩类的电致变色聚合物的光谱电化学图谱。
图7为本发明实施例7合成得到的新型噻吩并噻吩类的电致变色聚合物的方波函数图。
具体实施方式
本发明提供一种噻吩并噻吩类的电致变色聚合物及其制备方法,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明实施方式提供一种噻吩并噻吩类的电致变色聚合物,其分子结构通式为:
在本实施例中,通过在噻吩并[3,2-b]噻吩结构的3,6位点上引入具有强离子导电特征的寡聚醚侧链,并调节寡聚醚链的长度,形成噻吩并噻吩类单体;然后将所述噻吩并噻吩类单体与不同的双溴代单体共聚,可制得所述噻吩并噻吩类的电致变色聚合物;通过调节噻吩并噻吩类单体与双溴代单体的聚合度,可调节制得的电致变色聚合物的分子量、π共轭程度和分子链内及链间电荷转移能力,从而使得所述噻吩并噻吩类的电致变色聚合物具有高离子导电性、响应速率高、对比度高,且可以实现从多种颜色到无色之间的长期循环稳定性等特点。
在一些实施方式中,所述ar选自
在一些实施方式中,所述噻吩并噻吩类的电致变色聚合物为如下所示化学结构中的任意一种:
,其中,n为2-2000。
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些具体的实施方式中,所述噻吩并噻吩类的电致变色聚合物的分子结构式为:
在一些实施方式中,还提供一种噻吩并噻吩类的电致变色聚合物的制备方法,其中,包括步骤:
将噻吩并[3,2-b]噻吩、冰醋酸、氯仿和液溴混合搅拌,加热回流处理后,制得四溴噻吩并[3,2-b]噻吩化合物;
将锌单质与所述四溴噻吩并[3,2-b]噻吩化合物混合,再加入冰醋酸在室温条件下搅拌过夜,制得3,6-二溴噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将所述3,6-二溴噻吩并[3,2-b]噻吩化合物、溴化亚铜、碘化钾(41.47mg,0.25mmol)加入到甲醇钠溶液中,加热回流处理后,制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将所述3,6-二甲氧基噻吩并[3,2-b]噻吩化合物、对甲苯磺酸钠以及三乙二醇单乙醚加入甲苯溶剂中,加热回流处理后,制得分子结构式为
在氮气保护环境下,将化合物
本实施例通过一系列溴化反应、取代反应、直接芳基化偶联缩聚反应合成所述噻吩并噻吩类的电致变色聚合物,其合成方法简单方便,可重复性高,可控性强,产率高且不会对造成环境危害。所得电致变色聚合物具有高离子导电性,响应速率高、对比度高,且可以实现从多种颜色到无色之间的长期循环稳定性。
下面通过实施例对本发明进行详细说明:
实施例1
一种分子结构式为
其制备步骤如下所示:
1)、将噻吩并[3,2-b]噻吩(1.340g,10mmol)加入250ml的反应容器中,加入冰醋酸(20ml)和氯仿(100ml)混合溶剂,常温下逐滴滴入br2(4.735g,30mmol),再搅拌5小时,加热回流过夜;冷却至室温后,加入大量水,过滤洗涤,真空干燥得到四溴噻吩并[3,2-b]噻吩化合物3-1(4.065g,90%);
2)、在反应瓶中加入锌单质(1.151g,18mmol)和四溴噻吩并[3,2-b]噻吩化合物3-1(4.065g,9mmol),再加入冰醋酸(20ml),室温搅拌过夜;加入大量水,过滤洗涤,真空干燥得到3,6-二溴噻吩并[3,2-b]噻吩化合物3-2(2.514g,94.4%);
3)、在氮气保护环境下,将3,6-二溴噻吩并[3,2-b]噻吩化合物3-2(2.514g,8.5mmol),溴化亚铜(1.342g,17mmol),碘化钾(41.47mg,0.25mmol)加入含有甲醇钠(1.836g,34mmol)的甲醇溶剂(100ml)中,加入回流15小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到3,6-二甲氧基噻吩并[3,2-b]噻吩化合物3-3(1.6g,94%);
4)、在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物3-3(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物3-m1(1.77g,70%);
5)、在氮气保护环境下,将化合物3-m1(158.04mg,0.5mmol)、1,4-二溴苯(116.935mg,0.5mmol)、碳酸钾(138.21,mg,1mmol)、醋酸钯(4.49mg,0.02mmol)加入n,n-二甲基乙酰胺(5ml)溶剂中,120℃加热回流48小时;冷却至室温后将反应液滴入甲醇溶剂中析出固体,过滤得到粗产品;依次用甲醇、丙酮、正己烷、氯仿溶剂进行索氏提取,收集氯仿部分洗涤液,旋干得到噻吩并噻吩类的电致变色聚合物3(334mg,92%);
将本实施例1制备的噻吩并噻吩类的电致变色聚合物3溶解于氯仿溶剂中,喷涂到ito玻璃基底上,干燥得聚合物薄膜;以正四丁基六氟磷酸铵为电解质,乙腈为溶剂,采用三电极体系,喷涂在ito上聚合物薄膜作为工作电极,铂丝作为对电极,银丝作为参比电极;进行光谱电化学的测定,得到光谱电化学图谱(如图1所示)与方波函数图(如图2所示)。聚合物薄膜的变色性能表明,本实施例的聚合物薄膜可以从中性态的红色变为氧化态的无色,一些电致变色性能的参数见表1。
表1电致变色性能
由表1可知,本实施例1制备的新型噻吩并噻吩类的电致变色聚合物对比度高、响应时间短、着色效率高、可以实现从红色到无色之间稳定循环10000次而基本不衰减。
实施例2
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物15-m1;
在氮气保护环境下,将化合物15-m1(158.04mg,0.5mmol)、
实施例3
一种分子结构式为
,其制备方法包括步骤:
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物34-m1;
在氮气保护环境下,将化合物34-m1(158.04mg,0.5mmol)、
实施例4
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物51-m1;
在氮气保护环境下,将化合物51-m1(158.04mg,0.5mmol)、
实施例5
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物71-m1;
在氮气保护环境下,将化合物71-m1(158.04mg,0.5mmol)、
表2实施例5中聚合物薄膜的电致变色性能
实施例6
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物85-m1;
在氮气保护环境下,将化合物85-m1(158.04mg,0.5mmol)、
实施例7
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物96-m1;
在氮气保护环境下,将化合物96-m1(158.04mg,0.5mmol)、
表3实施例7中聚合物薄膜的电致变色性能
实施例8
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物98-m1;
在氮气保护环境下,将化合物98-m1(158.04mg,0.5mmol)、
实施例9
一种分子结构式为
,其制备方法包括步骤:
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物126-m1;
在氮气保护环境下,将化合物126-m1(158.04mg,0.5mmol)、
实施例10
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物135-m1;
在氮气保护环境下,将化合物135-m1(158.04mg,0.5mmol)、
实施例11
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物160-m1;
在氮气保护环境下,将化合物160-m1(158.04mg,0.5mmol)、
实施例12
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物192-m1;
在氮气保护环境下,将化合物192-m1(158.04mg,0.5mmol)、
实施例13
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物202-m1;
在氮气保护环境下,将化合物202-m1(158.04mg,0.5mmol)
实施例14
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物210-m1;
在氮气保护环境下,将化合物210-m1(158.04mg,0.5mmol)、
实施例15
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物223-m1;
在氮气保护环境下,将化合物223-m1(158.04mg,0.5mmol)、
实施例16
一种分子结构式为
根据实施例1中的制备方法制得3,6-二甲氧基噻吩并[3,2-b]噻吩化合物;
在氮气保护环境下,将3,6-二甲氧基噻吩并[3,2-b]噻吩化合物(1.6g,8mmol),对甲苯磺酸钠(0.275g,1.6mmol)、三乙二醇单乙醚(5.70g,32mmol)加入甲苯溶剂(80ml)中,加热回流20小时;冷却至室温后加入水,用二氯甲烷溶剂(300ml)萃取并洗涤多次,有机相干燥后加入硅胶旋干,柱层析分离得到化合物246-m1;
在氮气保护环境下,将化合物246-m1(158.04mg,0.5mmol)、
综上所述,本发明通过在噻吩并噻吩结构的3,6位点引入寡聚醚侧链,并调节寡聚醚链的长度;再将噻吩并噻吩类的单体与不同的双溴代单体通过直接芳基化偶联缩聚反应共聚,以调节材料的分子量、π共轭程度和分子链内及链间电荷转移能力。本发明提供的电致变色聚合物材料具有高离子导电性,响应速率高、对比度高,且可以实现从多种颜色到无色之间的长期循环稳定性。
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!