雪菊中黄诺玛苷和马里苷的提取方法与流程
本发明属于黄诺玛苷技术领域,具体涉及雪菊中黄诺玛苷和马里苷的提取方法。
背景技术:
昆仑雪菊(coreopsistinctoria)学名两色金鸡菊,为菊科(compositae)金鸡菊属(coreopsis)一年生草本植物。两色金鸡菊是原产于北美的观赏植物,我国引种成功后,在新疆有大规模种植,俗称雪菊,在当地被当作花茶饮用,维吾尔民间用来预防心血管疾病。在葡萄牙有每天喝两杯热两色金鸡菊茶饮用于控制糖尿病的传统。两色金鸡菊在新疆人们称之为雪菊、昆仑雪菊、昆仑血菊、高寒雪菊等,现已经逐渐作为一种茶饮普及开来。
国内外对两色金鸡菊的研究主要集中在黄酮类物质,发现两色金鸡菊中黄酮类物质是其药理活性成分。黄酮类化合物在植物界中广泛存在,并且许多都具有很好的生物活性,对人体健康有很多益处,能够保护人体不受疾病侵害。国内外对两色金鸡菊的研究主要集中在黄酮类物质,发现两色金鸡菊中黄酮类物质是其药理活性成分,其黄酮类物质主要包括查尔酮、二氢黄酮和橙酮等。两色金鸡菊头状花序中富含黄酮类成分,以查尔酮类和二氢黄酮类成分为其主要成分。植物化学研究已经证实,两色金鸡菊具有丰富的营养成分,目前已报道的从两色金鸡菊中分离鉴定出的29个黄酮类成分,这类化合物的母核包括黄酮类、二氢黄酮类、二氢黄酮醇类、查尔酮类、黄酮醇类、橙酮类,和黄烷醇类。课题组前期研究发现,雪菊中的主要成分有黄诺玛苷、马里苷、奥卡宁、黄酮奥卡宁等,并且可以通过hplc指纹图谱对其进行指认。
其中,黄诺玛苷可以增加高糖损伤的huvec(人皮静脉内皮细胞)的活力,改善糖毒性造成的细胞炎症反应,实现降低高糖诱导的皮内细胞损伤,起到保护内皮细胞的作用,从而延缓dr(糖尿病视网膜病变)的发展。马里苷具有抗病毒和抗糖尿病等作用。因此,可从雪菊中提取出黄诺玛苷和马里苷。但是,由于雪菊中有效成分种类较多,难以单独分离出某一单一物质,分离出的物质纯度较低,导致黄诺玛苷和马里苷的提取难度大,纯度低。
有鉴于此,有必要提出一种雪菊中黄诺玛苷和马里苷的提取方法,该方法可以提取出纯度较高的黄诺玛苷和马里苷。
技术实现要素:
本发明的目的在于提供一种雪菊中黄诺玛苷和马里苷的提取方法,该方法可以提取出纯度较高的黄诺玛苷。
为了实现上述目的,所采用的技术方案为:
雪菊中黄诺玛苷和马里苷的提取方法,包括以下步骤:
(1)将干燥的雪菊花序用乙醇溶液回流提取,得雪菊醇提物;
(2)将雪菊醇提物溶解于水中后,用乙酸乙酯萃取2次,收集水层,得萃余液;
(3)将萃余液用正丁醇萃取2次,收集有机层,得萃取液;
(4)将萃取液制成浸膏后,湿法上样,进行聚酰胺柱层析;
其中,所述聚酰胺柱层析采用梯度洗脱,流动相a为水,流动相b为乙醇,其体积分数变化依次为a相100%,b相0;a相90%,b相10%;a相80%,b相20%;a相60%,b相40%;a相50%,b相50%;a相5%,b相95%;
(5)收集采用a相90%,b相10%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的黄诺玛苷;
(6)收集采用a相60%,b相40%洗脱时的洗脱液,进行旋蒸,产生黄色结晶,得所述的马里苷。
进一步的,所述的步骤(1)中,采用质量分数为55%的乙醇溶液进行回流提取。
再进一步的,所述的回流提取的温度为47℃,时间为2.5h。
进一步的,所述的步骤(2)中,雪菊醇提物与水的质量体积比为10-15:200g/ml。
进一步的,所述的步骤(2)中的萃取过程为等体积萃取。
进一步的,所述的步骤(3)中的萃取过程为等体积萃取。
进一步的,所述的步骤(4)中,萃取液采用减压浓缩的方式制成浸膏。
进一步的,所述的步骤(4)中,梯度洗脱的流速为60ml/min。
进一步的,所述的步骤(4)中,聚酰胺的目数为60-100目。
进一步的,所述步骤(5)还包括:将收集的淡黄色结晶进行重结晶处理,得到所述的黄诺玛苷;
所述步骤(6)还包括:将收集的淡黄色结晶进行重结晶处理,得到所述的马里苷。
与现有技术相比,本发明的有益效果在于:
本发明所述的雪菊中黄诺玛苷和马里苷的提取方法,该方法通过萃取、聚酰胺柱柱层析,可以有效提取出黄诺玛苷和马里苷,且黄诺玛苷纯度可达93%以上,经过重结晶后,纯度可达98%以上;还有马里苷纯度可达91%以上,经过重结晶后,纯度可达98%以上。
附图说明
图1为雪菊醇提物指纹图谱;
图2为实施例1的工艺流程图;
图3为实施例1中雪菊聚酰胺柱层析液相色谱图(10%乙醇洗脱部位);
图4为实施例1中雪菊聚酰胺柱层析液相色谱图(40%乙醇洗脱部位);
图5为实施例2中雪菊聚酰胺柱层析液相色谱图(10%乙醇洗脱部位);
图6为实施例2中雪菊聚酰胺柱层析液相色谱图(40%乙醇部位洗脱)
图7为实施例3中雪菊聚酰胺柱层析液相色谱图(10%乙醇洗脱部位);
图8为实施例3中雪菊聚酰胺柱层析液相色谱图(40%乙醇洗脱部位);
图9为实施例4中雪菊硅胶柱层析黄诺玛苷重结晶液相色谱图;
图10为实施例4中雪菊硅胶柱层析马里苷重结晶液相色谱图;
图11为黄诺玛苷结构式;
图12为实施例4制备的黄诺玛苷红外吸收光谱图;
图13为实施例4制备的黄诺玛苷在esi(+)模式下的ms质谱图;
图14为实施例4制备的黄诺玛苷在esi(+)模式下的ms2二级质谱图;
图15为黄诺玛苷裂解示意图;
图16为实施例4制备的黄诺玛苷的1h-nmr(cd3od)谱。
图17为马里苷结构式;
图18为实施例4制备的马里苷在esi(+)模式下的ms质谱图;
图19为实施例4制备的马里苷红外吸收光谱图。
具体实施方式
为了进一步阐述本发明雪菊中黄诺玛苷和马里苷的提取方法,达到预期发明目的,以下结合较佳实施例,对依据本发明提出的雪菊中黄诺玛苷和马里苷的提取方法,其具体实施方式、结构、特征及其功效,详细说明如后。在下述说明中,不同的“一实施例”或“实施例”指的不一定是同一实施例。此外,一或多个实施例中的特定特征、结构或特点可由任何合适形式组合。
在详细阐述本发明雪菊中黄诺玛苷和马里苷的提取方法之前,有必要对本发明中提及的原料和方法等做进一步说明,以达到更好的效果。
2-apb的分子式为c14h16bno,分子量为225.1,化学名为2-氨基乙酯二苯基硼酸,是ins(1,4,5)p3诱导ca2+释放的拮抗剂,抑制ip3诱导的ca2+释放,ic50为42μm,但不影响ip3与其受体的结合。随着2-apb浓度增加(大于90μm),其抑制ca2+释放的作用也会相应增强。2-apb抑制ip3r,从而抑制钙库诱导的钙库调控性离子通道(soc)活化,以及受体诱导的trp3活化,ec50值通常在微摩尔范围。2-apb还可以有效地调控trp离子通道,阻断trpc1,trpc3,trpc5,trpc6,trpv6,trpm3,trpm7,trpm8和trpp2。在缺乏其他刺激因素时,2-apb可以刺激trpv1,trpv2和trpv3。2-apb选择性且特异性地阻断某些缝隙连接通道亚型。本发明实施例中采用的为sigma公司生产的2-apb。
聚乙二醇400,简称peg-400,为环氧乙烷和水开环聚合而成的混合物,分子式以ho(ch2ch2o)nh表示,其中n代表氧乙烯基的平均数。peg-400为无色或几乎无色的黏稠液体;略有特臭,在水或乙醇中易溶,在乙醚中不溶。本发明实施例中采用的为天津市百世化工有限公司生产的聚乙二醇400。
本发明实施例中采用的仪器有:
全息制备色谱(buchi,瑞士);中低压制备色谱(buchi,瑞士);点样仪(camaglinomat5);薄层色谱数码成像系统(camagreprostar3);双槽展开缸(20cm×10cm);硅胶g高效板(青岛海洋化工厂);高效液相色谱仪(日本岛津,lc-20ab泵,spd-20a紫外双波长检测器)。
本发明中未提及的工艺及工艺操作,为本领域的常规技术手段。
在了解了上述原料和方法等之后,下面将结合具体实施例对本发明雪菊中黄诺玛苷和马里苷的提取方法做进一步的详细介绍:
一实施例
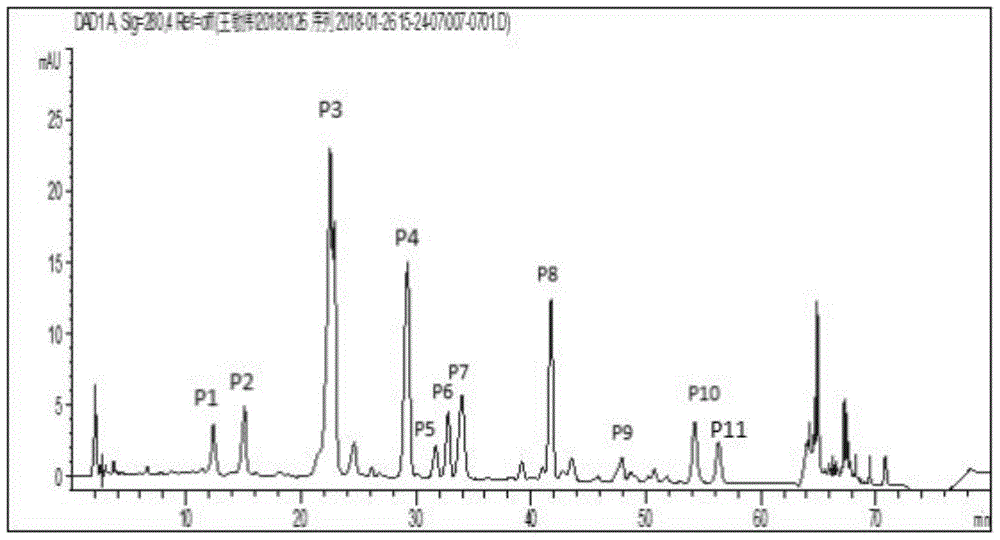
采用质量分数为55%的乙醇溶液得到的雪菊醇提物进行hplc分析。
hplc分析条件:shim-packvp-ods色谱柱(150mm×4.6mm,5μm);流速1.0ml/min;检测波长280nm;柱温:35℃;进样量10μl;流动相a:0.5%甲酸水;流动相b:乙腈,梯度洗脱程序如表1。
表1梯度洗脱程序
得到的醇提物指纹图谱如图1所示,图中的编号对应的成分如表2所示。
表2醇提物含有的成分
由图1和表2可知,雪菊醇提物中含有黄诺玛苷和马里苷。
实施例1.
其工艺路线如图2所示,具体操作步骤如下:
(1)将干燥的雪菊花序用质量分数为55%的乙醇溶液在47℃下,回流提取2.5h,得雪菊醇提物。
(2)将10g雪菊醇提物溶解于200ml水中后,用等体积的乙酸乙酯萃取,收集水层部分;然后用等体积的乙酸乙酯对水层部分萃取,再收集水层,得萃余液;
(3)将萃余液用等体积的正丁醇萃取,收集有机层部分;然后用等体积的正丁醇对有机层部分萃取,再收集有机层,即正丁醇部分,得萃取液。
(4)将萃取液减压浓缩后制成浸膏,湿法上样,进行聚酰胺柱层析(聚酰胺50g,60-100目;玻璃柱柱长:60cm,直径:4cm);
其中,所述聚酰胺柱层析采用梯度洗脱,流速为60ml/min,流动相a为水,流动相b为乙醇,每一个梯度冲3-5个柱体积,其体积分数变化如表2所示;
表2
(5)收集采用a相90%,b相10%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的黄诺玛苷。
(6)收集采用a相60%,b相40%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的马里苷。
分析:将上述黄诺玛苷结晶和马里苷结晶用水冲洗后,各称取4mg分别溶于4ml甲醇,经hplc分析,初步得知该结晶为黄诺玛苷和马里苷。通过面积归一化法,得知黄诺玛苷纯度为93.213%,马里苷初度为91.324%。
将步骤(5)收集的10%乙醇洗脱液进行hplc,得到的色谱图如图3所示,由图3可知,主要含有黄诺玛苷。
将步骤(6)收集的40%乙醇和洗脱液进行hplc,得到的色谱图如图4所示,由图4可知,主要含有马里苷。
本发明所述的雪菊中黄诺玛苷和马里苷的提取方法,该方法通过萃取、聚酰胺柱柱层析,可以有效提取出黄诺玛苷和马里苷,且纯度可达93.213%和91.324%。
实施例2.
其工艺路线如图2所示,具体操作步骤如下:
(1)将干燥的雪菊花序用质量分数为55%的乙醇溶液在47℃下,回流提取2.5h,得雪菊醇提物。
(2)将70g雪菊醇提物溶解于1000ml水中后,用等体积的乙酸乙酯萃取,收集水层部分;然后用等体积的乙酸乙酯对水层部分萃取,再收集水层,得萃余液;
(3)将萃余液用等体积的正丁醇萃取,收集有机层部分;然后用等体积的正丁醇对有机层部分萃取,再收集有机层,即正丁醇部分,得萃取液。
(4)将萃取液减压浓缩后制成浸膏,湿法上样,进行聚酰胺柱层析(聚酰胺300g,60-100目;玻璃柱柱长:60cm,直径:4cm);
其中,所述聚酰胺柱层析采用梯度洗脱,流速为60ml/min,流动相a为水,流动相b为乙醇,每一个梯度冲3-5个柱体积,其体积分数变化如表2所示。
(5)收集采用a相90%,b相10%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的黄诺玛苷。
(6)收集采用a相60%,b相40%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的马里苷。
分析:将上述黄诺玛苷结晶和马里苷用水冲洗后,各称取4mg分别溶于4ml甲醇,经hplc分析,初步得知该结晶为黄诺玛苷和马里苷,通过面积归一化法,得知其纯度为98.848%,马里苷结晶初度为93.751%。
将步骤(5)收集的10%乙醇洗脱液进行hplc,得到的色谱图如图5所示,由图5可知,主要含有黄诺玛苷。
将步骤(6)收集的40%乙醇洗脱液进行hplc,得到的色谱图如图6所示,由图6可知,主要含有马里苷。
本发明所述的雪菊中黄诺玛苷和马里苷的提取方法,该方法通过萃取、聚酰胺柱柱层析,可以有效提取出黄诺玛苷和马里苷,且纯度可达98.848%和93.751%。
实施例3.
其工艺路线如图2所示,具体操作步骤如下:
(1)将干燥的雪菊花序用质量分数为55%的乙醇溶液在47℃下,回流提取2.5h,得雪菊醇提物。
(2)将150g雪菊醇提物溶解于2000ml水中后,用等体积的乙酸乙酯萃取,收集水层部分;然后用等体积的乙酸乙酯对水层部分萃取,再收集水层,得萃余液;
(3)将萃余液用等体积的正丁醇萃取,收集有机层部分;然后用等体积的正丁醇对有机层部分萃取,再收集有机层,即正丁醇部分,得萃取液。
(4)将萃取液减压浓缩后制成浸膏,湿法上样,进行聚酰胺柱层析(聚酰胺600g,60-100目;玻璃柱柱长:60cm,直径:4cm);
其中,所述聚酰胺柱层析采用梯度洗脱,流速为60ml/min,流动相a为水,流动相b为乙醇,每一个梯度冲3-5个柱体积,其体积分数变化如表2所示。
(5)收集采用a相90%,b相10%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的黄诺玛苷。
(6)收集采用a相60%,b相40%洗脱时的洗脱液,进行旋蒸,产生淡黄色结晶,得所述的马里苷。
分析:将上述黄诺玛苷结晶和马里苷结晶用水冲洗后,各称取4mg分别溶于4ml甲醇,经hplc分析,初步得知该结晶分别为黄诺玛苷和马里苷,通过面积归一化法,得知黄诺玛苷纯度为97.151%,马里苷纯度为95.386%。
将步骤(5)收集的10%乙醇洗脱液进行hplc,得到的色谱图如图7所示,由图7可知,主要含有黄诺玛苷。
将步骤(6)收集的40%乙醇洗脱液进行hplc,得到的色谱图如图8所示,由图8可知,主要含有马里苷。
本发明所述的雪菊中黄诺玛苷和马里苷的提取方法,该方法通过萃取、聚酰胺柱柱层析,可以有效提取出黄诺玛苷和马里苷,且纯度可达97.151%和95.386%。
实施例4.
具体操作步骤如下:
将实施例1得到的黄诺玛苷及旋蒸后的母液合并后抽滤,放置于培养皿中阴干。取560mg溶于15ml的甲醇,静置一天,自然阴干,静置一天,有白色晶体析出。抽滤后,放置于培养皿中阴干,称重,共得110.25mg黄诺玛苷晶体。
将实施例1得到的马里苷及旋蒸后的母液合并后抽滤,放置于培养皿中阴干。取1.2g溶于40ml的甲醇,静置一天,自然阴干,静置一天,有橘黄色晶体析出。抽滤后,放置于培养皿中阴干,称重,共得510.25mg橘黄色晶体。
分析:取黄诺玛苷结晶溶于甲醇,制成浓度为1mg/ml的黄诺玛苷溶液。通过面积归一化法,得知其纯度为98.83%。
取马里苷结晶溶于甲醇,制成浓度为1mg/ml的黄诺玛苷溶液。通过面积归一化法,得知其纯度为98.552%。
将重结晶后的黄诺玛苷结晶进行hplc,得到的色谱图如图9所示,由图9可知,主要含有黄诺玛苷。
将重结晶后的马里苷结晶进行hplc,得到的色谱图如图10所示,由图10可知,主要含有马里苷。
本发明所述的雪菊中黄诺玛苷和马里苷的提取方法,该方法通过萃取、聚酰胺柱柱层析和重结晶,可以有效提取出黄诺玛苷和马里苷,且纯度可达98.83%和98.552%。
二实验测试
(1)黄诺玛苷
1、红外分析
(1)仪器:红外光谱仪(岛津ir-prestige,日本岛津)。
(2)实验方法:kbr压片法制样,将实施例4制备的黄诺玛苷晶体干燥、粉碎过200目筛,取样品粉末约2mg,分别与200mg溴化钾混合研磨充分均匀,压片测定,获得一维红外光谱图。
(3)结果:图11为黄诺玛苷结构式,图12为红外吸收光谱图,表3为红外吸收光谱归属。
表3黄诺玛苷外吸收光谱归属
结果分析:根据红外图谱可知3344cm-1与-oh的伸缩振动相符,表明结构中含有-oh;1670cm-1与c=o的伸缩振动相符,表明结构中含有c=o;1618-1521cm-1与苯环的伸缩振动相符,表明结构中含有苯环,1460-1321与ch2的伸缩振动相符,表明结构中含有ch2,1265与c-c的伸缩振动相符,表明结构中含有c-c,1082与c-o的伸缩振动相符,表明结构中含有c-o。本品的ir吸收光谱特征与黄诺玛苷结构相符。
2、质谱分析
(1)仪器:质谱仪(
(2)测定方法:称取实施例4制备的黄诺玛苷适量,加甲醇溶解,制成浓度约为1μg/ml的溶液作为样品溶液供测试用,分别测定供试品的一级和二级质谱图。
电离方式为电喷雾电离源(esi),多反应监测扫描(mrm),毛细管电压3kv,锥孔电压46v,离子源温度120℃,去溶温度350℃,去溶剂化气流:500l·hr-1,锥孔气流:50l·hr-1。
(3)结果与解析:质谱扫描范围m/z100-500,准分子离子峰[m-h]-m/z=449,提示分子量为449,与黄诺玛苷结构吻合;二级质谱的主要离子为135,151,287,(图13-14),按黄诺玛苷的结构对其碎片进行归属,能得到合理的解释(图15)。
3、核磁共振氢谱
(1)仪器:核磁共振光谱分析仪(inova600型,美国varian公司)
(2)测定方法:取实施例4制备的黄诺玛苷5mg溶于0.5mldmso(cd3od,99.9%)中供测试用。测定黄诺玛苷的1h-nmr。
(3)结果表明,黄诺玛苷的1h-nmr(cd3od)谱如图16所示,1h-nmr(600mhz,dmso)δ:9.08-9.04(2h,m,h-15,32),8.56(1h,s,h-32),8.54(1h,s,h-30),7.23-6.86(1h,m,h-28,14,27),6.76(1h,dd,j=1.9,1.0hz,h-24),5.44(1h,tt,j=6.2,1.1hzh-21),5.14-5.09(1h,m,h-11),4.84-4.81(1h,m,h-5),4.64-4.62(1h,m,h-10),4.12(1h,s,h-29),3.71-3.70(1h,m,h-12),4.22(1h,t,j=6.0hz,h-8);3.37(1h,ddd,j=12.4,7.6,5.4hz,h-7),3.16-3.13(3h,m,h-3,4,5),3.13-3.11(1h,m,h-1),2.68(1h,dd,j=15.4,7.1hz,h-20)。1h-nmr(600mhz,dmso)谱表明,除去活泼氢共振峰外,与黄诺玛苷的分子结构中氢原子组成相符。
4、熔点测定
(1)仪器:显微熔点仪(sgwx-4申光,上海精密科学仪器有限公司)。
(2)测定方法:调节升温速率使每分钟上升2.5-3.0℃,以样品开始产生气泡时的温度作为初熔温度,样品分解物开始膨胀上升时的温度为全熔温度。
(3)结果与分析:供试品经鼓风干燥箱80℃干燥30min,取少量黄诺玛苷于载玻片上,盖上盖玻片,放置在显微熔点仪载物台中央,开关调至加热状态,使温度不断升高,升温速率调至1-2℃/min,观察到黄诺玛苷由白色固体变为液态,测得奥卡宁熔点如表4所示,为253-254℃。
表4显微熔点仪测得黄诺玛苷熔点
测定黄诺玛苷之前,用酚酞标准品对显微熔点仪进行校正,其修正值为-2.2℃。
测定黄诺玛苷经过校正后其熔点为252.0-253.7℃,本实验测得值与文献报道黄诺玛苷熔点为253-254℃接近。
(2)马里苷
1、质谱分析
仪器:质谱仪(
测定方法:称取实施例4制备的马里苷适量,加甲醇溶解,制成浓度约为1μg/ml的溶液作为样品溶液供测试用,分别测定供试品的一级和二级质谱图。
电离方式为电喷雾电离源(esi),多反应监测扫描(mrm),毛细管电压3kv,锥孔电压46v,离子源温度120℃,去溶温度350℃,去溶剂化气流:500l·hr-1,锥孔气流:50l·hr-1。
结果与解析:质谱扫描范围m/z100-500,准分子离子峰[m-h]-m/z=449,提示分子量为449,与马里苷结构吻合;二级质谱的主要离子为135,151,287,(图18),按橙红色结晶的结构对其碎片进行归属,能得到合理的解释
2、红外分析
仪器:红外光谱仪(岛津ir-prestige,日本岛津)。
实验方法:kbr压片法制样,将实施例4制备的马里苷晶体干燥、粉碎过200目筛,取样品粉末约2mg,分别与200mg溴化钾混合研磨充分均匀,压片测定,获得一维红外光谱图。
结果分析:图17为马里苷结构式,根据红外图谱(图19)可知,3271.27cm-1与-oh的伸缩振动相符,表明结构中含有-oh;1633.71cm-1与c=o的伸缩振动相符,表明结构中含有c=o;1618-1521cm-1与苯环的伸缩振动相符,表明结构中含有苯环,1273.02与c-c的伸缩振动相符,表明结构中含有c-c,1076.28与c-o的伸缩振动相符,表明结构中含有c-o。本品的ir吸收光谱特征与马里苷结构相符。
以上所述,仅是本发明实施例的较佳实施例而已,并非对本发明实施例作任何形式上的限制,依据本发明实施例的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明实施例技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!