一种内源性酶触发的DNA步行器及其检测UDG的应用的制作方法
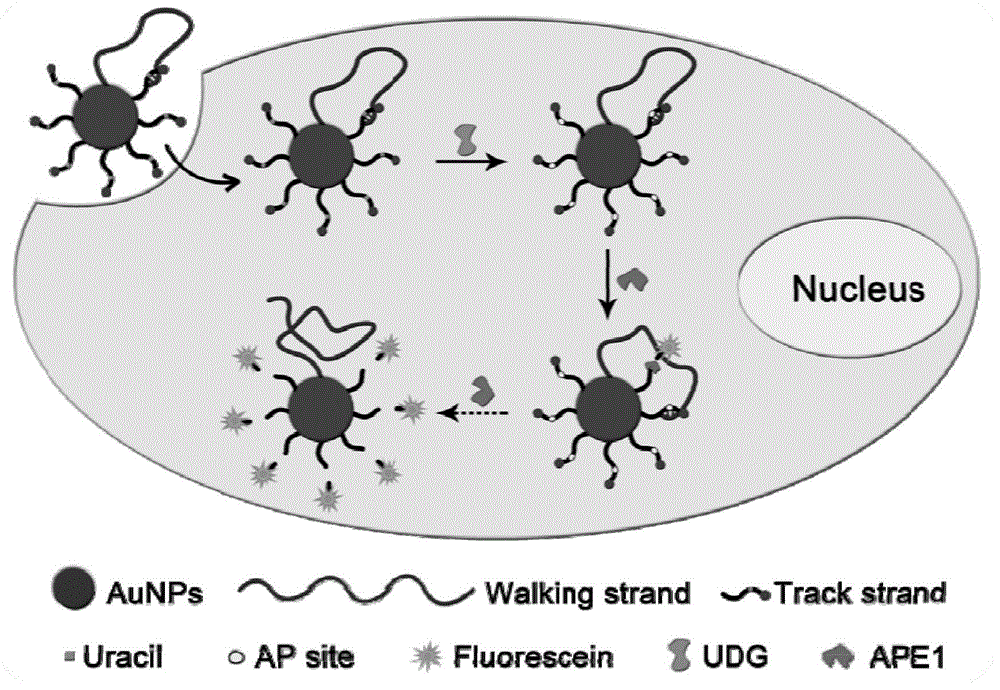
本公开涉及一种内源性酶触发的dna步行器及其检测udg的应用。
背景技术:
这里的陈述仅提供与本公开有关的背景信息,而不必然构成现有技术。
尿嘧啶-dna糖基化酶(udg)是一种dna损伤修复蛋白,其主要功能是通过催化尿嘧啶和脱氧核糖之间的n-糖苷键水解来去除尿嘧啶。udg的功能是尿嘧啶碱基切除修复(uber)的第一步,然后是脱嘌呤/脱嘧啶(ap)内切核酸酶,连接酶和聚合酶作用以完成uber途径,维持基因组的完整性。然而,细胞内udg活性异常会导致dna复制过程中错误引入尿嘧啶,导致不正确的碱基配对和基因突变,最终增加疾病的发生率,例如免疫缺陷症、布鲁姆综合征和淋巴瘤。细胞内udg活性检测对于深入理解dna修复系统和进一步研究突变相关的疾病至关重要。dna探针被细胞内核酸酶降解导致高背景并干扰检测灵敏度,从而使得udg的检测较为困难。
dna分子机器是由合成寡核苷酸组成的特殊设计的组件,它们改变状态并进行机器式运动以响应外部刺激。它类似于天然生物分子机器,例如通过一系列能量转换步骤和合作行为参与生命系统中重要过程的运动蛋白。与蛋白质相比,dna的独特性质,包括碱基配对的高保真性,结构和相互作用的简单性,使其成为构建可控分子机器的有前途的材料。已经报道了各种dna分子机器,包括步行器、镊子、齿轮、旋转器、节拍器和转运器,它们开始在传感,程序合成和器件制造中显示出有吸引力的应用。dna步行器是一类独特的dna分子机器,可使特殊组分寡核苷酸沿着预先设计的轨道逐步移动。dna步行器的特点是它可以在特殊刺激的驱动下自主且连续地行走多步。它可以是固有信号放大器,因为重复步骤可以引起反应循环以产生累积信号。大多数dna步行器通过体外刺激在胞外运行。据本公开发明人所知,最近,研究人员开始探索在活细胞中运行的dna步行器,其行为与天然生物分子机器类似。然而,经本公开发明人研究发现,为了维持胞内dna步行器的运行,须引入外源金属离子(mn2+,zn2+)或燃料寡核苷酸,这会导致细胞状态的改变或完整燃料dna的降解,最终限制细胞中dna步行器的运行,由于dna链对非特异性核酸酶降解的抵抗和步行器独特的扩增能力和纳米颗粒3-d表面上强大的dna富集能力,dna步行器在活细胞中运行成像udg活性,而udg活性的成像之前存在未受保护的dna探针以及细胞内扩增的困难引起的灵敏度有限的问题。
技术实现要素:
为了解决现有技术的不足,本公开的目的是提供一种内源性酶触发的dna步行器及其检测udg的应用,该dna步行器对udg的检测有良好的特异性及灵敏度。
为了实现上述目的,本公开的技术方案为:
一方面,一种内源性酶触发的dna步行器,包括金纳米颗粒、至少一条行走链和若干轨道链,所述行走链和轨道链均为单链dna,行走链的一端与金纳米颗粒连接,每条轨道链的一端均与金纳米颗粒连接,每条轨道链的另一端均连接荧光团,行走链含有第一序列,轨道链的含有第二序列,第一序列与第二序列互补,第二序列中的一个碱基为尿嘧啶碱基。
本公开将dna步行器构建在金纳米颗粒(aunp)表面上,由行走链和数倍的荧光团标记的含尿嘧啶的轨道链构成。荧光素的荧光被金纳米粒子的表面等离子体吸收并淬灭。行走链与轨道链部分互补并可杂交。通过纳米颗粒辅助的内吞作用,dna步行器,包括其所有成分,可以很容易地进入活细胞。此外,纳米颗粒表面固定的dna可以被保护免受核酸酶降解,以保持dna步行器的完整性并在胞内运行。在内源性udg的作用下,去除了轨道链中的尿嘧啶碱基,留下了ap位点。随后,在内源性脱嘌呤/脱嘧啶核酸内切酶(ape1)的作用下ap位点被切割,荧光素被释放并恢复荧光。然后,行走链继续与下一个轨道链杂交并启动下一个轨道链解离和荧光素释放循环,沿着三维(3-d)表面移动直到所有轨道链被消耗。
另一方面,一种上述dna步行器在检测udg中的应用。
第三方面,一种检测udg的生物传感器,包括上述dna步行器和脱嘌呤/脱嘧啶核酸内切酶。
第四方面,一种检测udg的检测试剂盒,包括上述dna步行器、脱嘌呤/脱嘧啶核酸内切酶、1×nebcutsmart缓冲液和pbs缓冲液。
第五方面,一种检测udg的方法,采用上述dna步行器、生物传感器或检测试剂盒,将dna步行器加入待测溶液中进行孵育,再添加脱嘌呤/脱嘧啶核酸内切酶进行反应,然后进行荧光检测。
本公开的有益效果为:
本公开的内源酶驱动的dna步行器可用于在活细胞中成像udg活性,在udg和ape1的作用下,杂交的含尿嘧啶的轨道链被去除碱基并切割,使得行走链的连续解离/杂交并沿着3-d轨道运动。携带荧光团的被切割的大量轨道链片段被释放以恢复荧光和放大信号输出。dna步行器具有明确定义的轨道,显示了对靶酶的良好特异性,具有对非特异性核酸酶降解的抗性,从而提高检测udg的灵敏度。并且本公开的dna步行器可以在活细胞中独立运行。
附图说明
构成本公开的一部分的说明书附图用来提供对本公开的进一步理解,本公开的示意性实施例及其说明用于解释本公开,并不构成对本公开的不当限定。
图1为本公开的原理图;
图2为本公开实施例制备的纳米金颗粒的结构表征图,a为紫外-可见吸收光谱,b为透射电子显微镜照片;
图3为本公开实施例中荧光强度与已知金纳米粒子表面已知浓度轨道链之间的校正曲线,插图为dtt孵育的dna步行器上清液的荧光光谱;
图4为本公开实施例中dna步行器和其dna-aunp类似物(将u碱基替换为t碱基)在1u/mludg和30u/mlape1存在时荧光强度随时间的变化曲线;
图5为本公开实施例中行走链和轨道链间互补碱基数目的优化表征柱状图,fwalker和fcontrol分别代表dna步行器和其dna-aunp类似物(将u碱基替换为t碱基)在1u/mludg和30u/mlape1作用下所产生的荧光强度;
图6为本公开实施例dna步行器的表征图,a为dna步行器和其dna-aunp类似物在udg和ape1存在时的荧光光谱,b为非变性聚丙烯酰胺凝胶电泳表征图,泳道m为dnamarker,泳道1为巯基化的轨道链,泳道2为巯基化的行走链,泳道3为dna步行器运行结束后取代下的寡聚核酸链,泳道4为dna-aunp对照物运行后取代下的寡聚核酸链;
图7为本公开实施例dna步行器荧光增加的原理表征图,a为在1u/mludg和30u/mlape1作用下dna步行器与轨道链修饰金纳米对照物的荧光光谱,b为dna步行器与含有行走链修饰金纳米对照物/轨道链修饰金纳米对照物的混合物的荧光光谱,a为dnawalker,b为walkingstrand-aunp+trackstrand-aunp;
图8为本公开实施例dna步行器的特异性表征图,haag、hogg1、endoiv和udg的浓度均为1u/ml;
图9为本公开实施例中ugi对dna步行器运行的抑制作用的表征曲线;
图10为本公开实施例dna步行器在udg和ape1、dnasei和缓冲液作用下荧光强度随时间的变化曲线;
图11为本公开实施例使用dna步行器(上面一行)和其dna-aunp类似物(下面一行)荧光成像hela细胞中udg酶活性的结果表征图;
图12为本公开实施例对dna步行器和其dna-aunp类似物处理后的细胞进行流式细胞计数所得到的归一化平均荧光强度柱状图;
图13为本公开实施例对dna步行器对udg检测限的表征图,a为dna步行器在不同浓度udg(0、0.1u/ml、0.2u/ml、0.3u/ml、0.5u/ml、0.8u/ml、1u/ml)和ape1(30u/ml)作用下的荧光光谱,b为520nm处荧光强度与udg浓度之间的线性关系图。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本公开提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本公开所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本公开的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
鉴于现有技术难以对udg进行检测的不足,为了解决如上的技术问题,本公开提出了一种内源性酶触发的dna步行器及其检测udg的应用。
本公开的一种典型实施方式,提供了一种内源性酶触发的dna步行器,包括金纳米颗粒、至少一条行走链和若干轨道链,所述行走链和轨道链均为单链dna,行走链的一端与金纳米颗粒连接,每条轨道链的一端均与金纳米颗粒连接,每条轨道链的另一端均连接荧光团,行走链含有第一序列,轨道链的含有第二序列,第一序列与第二序列互补,第二序列中的一个碱基为尿嘧啶碱基。
本公开原理如图1所示,将dna步行器构建在金纳米颗粒(aunp)表面上,由行走链和数倍的荧光团标记的含尿嘧啶的轨道链构成。荧光素的荧光被金纳米粒子的表面等离子体吸收并淬灭。行走链与轨道链部分互补并可杂交。通过纳米颗粒辅助的内吞作用,dna步行器,包括其所有成分,可以很容易地进入活细胞。此外,纳米颗粒表面固定的dna可以被保护免受核酸酶降解,以保持dna步行器的完整性并在胞内运行。在内源性udg的作用下,去除了轨道链中的尿嘧啶碱基,留下了ap位点。随后,在内源性脱嘌呤/脱嘧啶核酸内切酶(ape1)的作用下ap位点被切割,荧光素被释放并恢复荧光。然后,行走链继续与下一个轨道链杂交并启动下一个轨道链解离和荧光素释放循环,沿着三维(3-d)表面移动直到所有轨道链被消耗。
该实施方式的一种或多种实施例中,第一序列的碱基个数为数目为8个。通过实验证明,当行走链和轨道链间互补碱基数目为8个时,具有更高的荧光强度,检测效果更好。
该实施方式的一种或多种实施例中,行走链和轨道链均通过au-s键与金纳米颗粒连接。能够增强检测效果。
该实施方式的一种或多种实施例中,所述荧光团为fam。
该实施方式的一种或多种实施例中,所述金纳米颗粒的粒径为12~14nm。
该实施方式的一种或多种实施例中,行走链和轨道链的摩尔比为1:15~25。
该实施方式的一种或多种实施例中,行走链从5’端至3’端的序列为:tttttttttttttttttttttttttttttttttttttttttttttttttttcctaagc,
轨道链从5’端至3’端的序列为:ttttttttttgctuaggat。采用该行走链和轨道链的检测效果更好。
本公开的另一种实施方式,提供了一种上述dna步行器在检测udg中的应用。本公开所述的应用以非疾病的诊断为目的。
本公开的第三种实施方式,提供了一种检测udg的生物传感器,包括上述dna步行器和脱嘌呤/脱嘧啶核酸内切酶。
本公开的第四种实施方式,提供了一种检测udg的检测试剂盒,包括上述dna步行器、脱嘌呤/脱嘧啶核酸内切酶、1×nebcutsmart缓冲液和pbs缓冲液。
本公开的第五种实施方式,提供了一种检测udg的方法,采用上述dna步行器、生物传感器或检测试剂盒,将dna步行器加入待测溶液中进行孵育,再添加脱嘌呤/脱嘧啶核酸内切酶进行反应,然后进行荧光检测。
该实施方式的一种或多种实施例中,所述孵育温度为36.5~37.5℃,孵育时间为1~1.5h。
该实施方式的一种或多种实施例中,反应后进行离心,取离心后的上清液进行荧光检测。
该实施方式的一种或多种实施例中,荧光检测中,激发波长为488nm,发射波长为520nm。
为了使得本领域技术人员能够更加清楚地了解本公开的技术方案,以下将结合具体的实施例详细说明本公开的技术方案。
实施例
1.实验部分
1.1试剂和原料
te缓冲溶液(10mmtris-hcl,ph8.0,1mmedta)制备用于稀释合成的dna粉末,然后储存在-20℃。13nm金纳米颗粒由三水合氯金酸,二水合柠檬酸钠制备而成。尿嘧啶-dna糖基化酶(udg)、尿嘧啶糖基化酶抑制剂(ugi)、8-氧化鸟嘌呤dna糖基化酶(hogg1)、人类烷基腺嘌呤dna糖基化酶(hagga)、内切酶iv(endoiv)和无碱基/嘌呤内切酶1(ape1)购自于新英格兰生物有限公司(北京,中国)。tween20购买自北京索莱宝科技有限公司。磷酸盐缓冲盐(8mmna2hpo4,2mmkh2po4,136mmnacl,2.6mmkcl,ph7.4)购自美国克伦威尔生物工业公司。一些分析纯的化学试剂无需纯化。所有溶液均是用超纯水(电阻率>18.25mωcm)制备的,超纯水是由milliporemilli-q水荧光化系统制得的。dna寡核苷酸链均购自于生工生物工程股份有限公司(上海,中国),见表1。
表1寡核苷酸序列
下划线的字母表示行走链和轨道链之间的互补碱基,阴影的字母表示尿嘧啶碱基或取代的胸腺嘧啶碱基。轨道链名称中的数字表示行走链和轨道链之间互补碱基的数量。根据紫外分光光度和序列消光系数测定寡核苷酸的浓度。
荧光测定的激发和发射波长参数分别为488nm和520nm。激发狭缝和发射狭缝均为10nm,光电倍增管电压为700v,扫描速度是240/s,响应时间为2s。
1.2.实验步骤
1.2.1制备13nm金纳米颗粒
aunps(粒径13nm)的合成采用柠檬酸钠还原氯金酸的方法。在250ml的三颈烧瓶中加入磁子和45ml纯水,在缓慢搅拌下加热至沸腾,此时调整磁子转速为剧烈搅拌,紧接着加入5ml10mm三水合氯金酸,2min后迅速加入5ml38.8mm柠檬酸纳,继续在剧烈加热搅拌下沸腾10min,然后关闭电热套,降低磁子转速,保持在缓慢搅拌状态下,自然冷却至室温,过滤备用。最后利用紫外分光光度计和电子显微镜(tem)表征aunps。摩尔吸光系数为2.7×108lmol-1cm-1)。
1.2.2制备dna步行器(dna-aunp探针)及dna修饰量表征
按照文献(juewenliu,yilu,preparationofaptamer-linkedgoldnanoparticlepurpleaggregatesforcolorimetricsensingofanalytes.nat.protoc.,2006,1,246-252.)报道的方法将dna探针修饰到13nmaunps上完成dna步行器的构建。巯基化的行走链和轨道链寡核苷酸按照1:20的比例混合修饰。2.0μl50μm行走链和20μl100μm轨道链寡核苷酸加入到500μl9nmaunps中。混合物室温下避光孵育过夜16h,然后加入50μl超纯水(含1%的tween20)孵育10min,其次缓慢加入80μl2mnacl,分8次加完,室温避光孵育12h。最后,溶液在12000rmp下30min来分离出来功能化dna链的aunps(dna步行器)。这些dna步行器用1×pbs缓冲液洗涤三次,最后用200μlpbs缓冲液稀释,放在4℃保存备用。当使用含有尿嘧啶的行走链和轨道链对aunps进行功能化时,获得了dna步行器;当使用含有胸腺嘧啶的行走链和轨道链对aunps进行功能化时,获得了dna步行器的对照类似物。其他对照dna-aunp类似物的制备方法还包括:仅用轨道链对aunp进行功能化,或使用行走链和聚胸腺嘧啶轨道链对aunp进行功能化。
修饰到aunps上的轨道链数量是根据文献(sarahj.hurst,abigailk.r.lytton-jean,c.a.mirkin,maximizingdnaloadingonarangeofgoldnanoparticlesizes,anal.chem.,2006,78,8313-8318.)报道的方法进行定量的。将一定浓度的dna修饰aunps探针与二硫苏糖醇(dtt,终浓度为20mm)混匀,在室温震荡的条件下避光孵育过夜,aunps表面上的dna链可以被解离下来。然后离心,并利用荧光分光光度计测定解离下来的标记fam的dna链在520nm处的荧光强度。接着在溶液组成相同的条件下,测定一系列浓度的标记fam的轨道链在520nm处的荧光强度,并绘制标准曲线。通过解离下来的轨道链的荧光强度和标准曲线的线性方程计算出解离下来的轨道链的浓度,用此浓度除以dna步行器探针浓度即可估算出每个探针上结合的轨道链数量。
1.2.3分析dna步行器可行性
将40μl的含有0.5nmdna步行器、1u/mludg和1×nebcutsmart缓冲液的混合物在37℃下孵育1h后,接着添加30u/mlape1反应1h,最后在12000rmp下离心30min,取上清液进行荧光测量。激发和发射波长分别采用488nm、520nm。对照组中使用含有胸腺嘧啶的dna-aunps类似物,步骤同上。
15%的非变性聚丙烯酰胺电泳(page)用于分析反应产物。1×tbe(89mmtris,89mmboricacid,2.0mmedta,ph8.3)用于电泳缓冲液。page在恒流为30ma下1.5h完成,0.7%(w/v)是在恒压200v下室温1h完成。sybrgold染色40min。照片由紫外线成像系统(bio-radlaboratories)获得。
1.2.4dna-aunps探针稳定性研究
将dnasei(2.5μu)和目标物分别与dna-aunps核酸探针(0.5nm)混和,然后在一定的时间分别测定荧光强度。
1.2.5dna-aunps核酸探针特异性分析
在含有0.5nmdna步行器、1×nebcutsmart缓冲液和pbs的混合溶液中分别加入适量udg、hagg、hogg1和endoiv,在37℃下孵育1h。接着添加ape1反应1h,最后在12,000rmp下离心30min,取上清液进行荧光测量。激发和发射波长分别采用488nm和520nm。
1.2.6udg抑制剂分析
加入探针之前,将udg与ugi混合两分钟,将反应物放置37℃反应1h,然后加入ape1反应1h,离心取上清液测定荧光强度。我们用相对荧光强度用于评价ugi对udg的抑制效果。以上所有的实验均重复三次,用三次实验结果的标准偏差表示误差棒。
1.2.7细胞培养
在1640培养基中加入10%胎牛血清的和1%的双抗作为培养液,培养环境是含有5%co2湿润的37℃培养箱中。
1.2.8dna-aunps核酸探针的细胞内荧光成像实验
将hela接种在共聚焦小皿上,贴壁生长24h后,分别加入dna步行器和该步行器的dna-aunp对照物(1nm),然后继续培养3.0h后,置于荧光显微镜下进行成像。
1.2.9细胞流式细胞术
将密度为400000个hela细胞/孔孵育在6孔板上,过夜孵育24h。次日,用pbs清洗细胞,去除抗生素和血清,并添加探针。细胞在37℃培养3h。用pbs冲洗一次后,用流式细胞仪对细胞进行分析。用激光激发488nm测量荧光。
2.表征结果
通过使用巯基化的58-mer行走链和19-mer含尿嘧啶的轨道链(表1)以1:20的链浓度功能化13nmaunp(aunp结构如图2所示)来制备dna助行器。通过二硫苏糖醇(dtt)-解离方法,如图3所示,确定轨道链的数量为每个aunp267链,表明轨道链在纳米颗粒表面上富集,用于多步行走和信号放大。dna步行器首先通过添加纯化酶udg和ape1的在胞外测试。不与udg相互作用的对照dna-aunp缀合物在其他条件相同的条件下制备,但用胸腺嘧啶碱基取代轨道链中的尿嘧啶碱基。dna步行器通过记录的时间依赖性荧光变化显示了高运行速率,如图4所示。步行链和轨道链之间的互补碱基数显着影响dna步行器的运行。观察到8个互补碱基是最佳的,如图5所示,较少的互补碱基可能降低杂交效率,过多的互补碱基可能使纳米粒子之间的dna杂交,这两者都会损害三维轨道上行走链的运动。
dna步行器在udg和ape1的作用下产生高荧光,如图6a所示,这是由于轨道链形成ap位点并且在步行链的移动过程中荧光团被切割释放。相反,如果将尿嘧啶碱基改变为胸腺嘧啶碱基,则荧光低4.3倍(图6a)。这是因为udg不能作用于胸腺嘧啶碱基,因此抑制了轨道链的切割和步进链的连续移动。由于纯化的酶储存在含有dtt的缓冲液中,因此少量的轨道链从aunp解离以产生背景荧光。在dna步行器运行之后,纳米颗粒表面上的dna链被dtt置换并通过天然聚丙烯酰胺凝胶电泳(page)表征。通过与dna标记带比较(图6b,泳道m、1和2),观察到在udg和ape1的作用下,对应于含有尿嘧啶的轨道链的条带可忽略不计(图6b,泳道3),表明轨道链被切割成对染色和成像不敏感的较短寡核苷酸。相反,如果将尿嘧啶碱基改变为胸腺嘧啶碱基(图6b,泳道4),则轨道链保持其完整性,这解释了低荧光并验证了运行的抑制。
为了证明荧光增加是否由于行走链的移动引起,制备了仅有功能化轨道链的对照dna-aunp缀合物。只能观察到对照缀合物的背景荧光,如图7a所示,表明行走链在dna步器者的运行中是必不可少的。如果行走链沿三维轨道移动还是纳米颗粒之间跳跃,制备了两种对照dna-aunp缀合物,分别在aunp上修饰功能化行走链和轨道链。特别是,硫醇化的等长的聚胸腺嘧啶链取代了轨道链,目的是使其行走链与原dna步行器表面覆盖率相似。在udg和ape1的作用下,两种dna和aunp缀合物的混合物产生的荧光比原dna步行器低4.2倍,如图7b所示,表明每个dna步行器的独立运行并沿其三维轨道行走。dna步行器特异性地响应刺激,因为它的启动是由udg特异性切除尿嘧啶引起的。它在udg的作用下运行,但是对其他糖基化酶如8-氧鸟嘌呤dna糖基化酶(hogg1)和人烷基腺嘌呤dna糖基化酶(haag)以及其他核酸内切酶如脱嘌呤/脱嘧啶核酸内切酶iv(内切iv)都没有响应,如图8所示。如图9所示,尿嘧啶糖基化酶抑制剂(ugi)以1:1摩尔量与udg结合形成紧密且生理学上不可逆的复合物,以剂量依赖性方式逐渐减少dna步行器的运行。如图10所示,经非特异性切割单链和双链dna的dnasei测试,dna步行器显示出对非特异性核酸酶降解的良好抗性。
之后在活细胞中测试dna步行器的运行。选择人宫颈癌(hela)细胞作为模型细胞,并将其接种在共聚焦培养皿上,生长时间为24小时。分别加入dna步行器及其对照dna-aunp缀合物,并与细胞一起温育3小时,以进行内化和运行。值得注意的是,不需要添加需固定细胞并影响细胞内物质活性的外源酶。结果通过荧光显微镜成像。观察到dna步行器在细胞内产生明显的绿色荧光信号(图11,第一行),表明在细胞内源性udg和ape1的作用下,步行链绕着aunp表面移动,并且轨道链被切割。释放大量的荧光素。相比之下,用胸腺嘧啶代替尿嘧啶的对照dna-aunp缀合物产生非常弱的荧光信号(图11,第二行),表明在细胞内荧光素标记的链不会从aunp表面降解或分离产生假阳性信号。因此,观察到的明亮荧光是由酶介导的轨道链断裂引起的,并且dna步行器可以在活细胞内运行良好。dna步行器的运行可以直接成像与人类细胞系相关的udg酶活性。
为了表征dna步行器在大量细胞中的运行,进一步进行流式细胞术实验。将hela细胞接种在孔板中,生长时间为24小时。分别加入dna步行器及其对照dna-aunp缀合物并孵育3小时。用磷酸盐缓冲盐水(pbs)洗涤后,计数1×104、4×104和8×104细胞样品,并根据细胞相关荧光强度分别用流式细胞仪分析。观察到在三个细胞组中dna步行器与对照dna-aunp缀合物(图12)相比显示明显的荧光增强,表明dna步行器在细胞内udg和ape1的作用下运行并释放荧光团。相比之下,用胸腺嘧啶替代尿嘧啶的对照dna-aunp缀合物产生类似于未处理细胞的背景荧光信号,如图12所示,这是由于内源酶不能作用于表面缀合的dna链并且不能驱动其运行。定量数据揭示了dna步行器相对于对照dna-aunp缀合物有平均3.7倍的荧光增强,并且两组之间的信号差别非常明显(p<0.01),表明dna步行器在细胞内的有效运行。
对udg酶检测限的表征,如图13所示。dna步行器随udg浓度的增加产生逐渐增强的荧光,且荧光强度与udg浓度在0.03到1u/ml的范围内呈现线性关系,检测限计算为0.03u/ml。
总之,本实施例证明了由内源酶驱动的dna步行器可用于在活细胞中成像udg活性。在细胞内udg和ape1的作用下,杂交的含尿嘧啶的轨道链被去除碱基并切割,使得行走链的连续解离/杂交并沿着3-d轨道运动。携带荧光团的被切割的大量轨道链片段被释放以恢复荧光和放大信号输出。dna步行器具有明确定义的轨道,显示了对靶酶的良好特异性,具有对非特异性核酸酶降解的抗性,并且可以在活细胞中独立运行。与将atp等小分子作为刺激不同,通过更复杂的蛋白质驱动的dna步行器能更多地模仿天然生物分子机器的先进生物学功能。由于纳米粒子表面结合的dna对非特异性核酸酶降解的抗性和独特的信号放大能力,构建的dna步行器能对细胞内的udg活性进行灵敏成像。
以上所述仅为本公开的优选实施例而已,并不用于限制本公开,对于本领域的技术人员来说,本公开可以有各种更改和变化。凡在本公开的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本公开的保护范围之内。
sequencelisting
<110>山东大学
<120>一种内源性酶触发的dna步行器及其检测udg的应用
<130>
<160>10
<170>patentinversion3.3
<210>1
<211>58
<212>dna
<213>人工序列
<400>1
tttttttttttttttttttttttttttttttttttttttttttttttttttcctaagc58
<210>2
<211>19
<212>dna
<213>人工序列
<400>2
ttttttttttgctuaggat19
<210>3
<211>19
<212>dna
<213>人工序列
<400>3
ttttttttttgcttaggat19
<210>4
<211>19
<212>dna
<213>人工序列
<400>4
ttttttttttttttttttt19
<210>5
<211>61
<212>dna
<213>人工序列
<400>5
ttttttttttttttttttttttttttttttttttttttttttttttttttatcctaagca60
a61
<210>6
<211>19
<212>dna
<213>人工序列
<400>6
ttttttttttgctuaggat19
<210>7
<211>19
<212>dna
<213>人工序列
<400>7
ttttttttttgcttaggat19
<210>8
<211>55
<212>dna
<213>人工序列
<400>8
ttttttttttttttttttttttttttttttttttttttttttttttttttctaag55
<210>9
<211>19
<212>dna
<213>人工序列
<400>9
ttttttttttgctuaggat19
<210>10
<211>19
<212>dna
<213>人工序列
<400>10
ttttttttttgcttaggat19
- 还没有人留言评论。精彩留言会获得点赞!