一种PARP抑制剂中间体的结晶方法与流程
本发明涉及一种parp抑制剂中间体的结晶方法,特别涉及pamiparib的中间体(r)-2-(1-(2-(叔丁基氧基)-2-氧代乙基)-2-甲基吡咯烷-2基)-6-氟-1-甲苯磺酰基-1h-吲哚-4-羧酸甲酯
背景技术:
wo2013/097225a1公开了作为聚(adp-核糖基)转移酶(parps)抑制剂,并具体公开了化合物
4(5h)-酮大规模合成的制备方法,其中,
bg-11是pamiparib生产过程中关键的质量控制点,然而bg-11是一种极性较小的化合物,并且其在多种极性溶剂中均具有良好的溶解度,例如,室温下在诸如乙腈,四氢呋喃,乙酸乙酯甚至甲基叔丁基醚中都有比较好的溶解度,这给化合物的结晶的溶剂选择纯化带来了一定困难。这些极性有机溶剂或者混合溶剂均不适合作为bg-11的结晶溶剂。然而,本发明人在进行大量筛选实验时发现当重结晶溶剂选择醇类溶剂,或者醇/水混合溶剂时,且在降温析晶的过程中将温度降低至20℃以下,将获得纯度极高的bg-11,该工艺大大提高了大规模生产过程中中间体bg-11的纯度,可以得到bg-11的粉末状晶体,物料性状好,产物稳定,在后续生产步骤中使用方便。该结晶方法除杂能力极强,能够基本完全去除前面步骤形成和携带的有关杂质物质,产品hplc(高效液相色谱)纯度接近100%。同时该结晶方法还能有效去除催化剂引入的钯和铜残留(典型批次中钯和铜残留可以分别控制到不超过10ppm),因此极大地提高了药品中间体bg-11的生产质量。
技术实现要素:
本发明涉及一种parp抑制剂pamiparib的中间体
(i)将化合物1粗品溶液加入极性溶剂1;所述极性溶剂1选自醇类溶剂或醇/水混合溶剂;
(ii)加热搅拌至澄清;
(iii)降温冷却,任选地加入晶种,加入水,析晶;
(iv)进一步降温并搅拌使晶态物质充分析出;所述的降温所至的温度为不高于20℃;
(v)分离晶态物质,用极性溶剂2/水混合液洗涤,任选地干燥所得物质,得到化合物1的结晶;
所述极性溶剂2选自醇类溶剂。
优选地,步骤(i)中所述的醇类溶剂选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇中的一种或多种;醇/水的混合溶剂选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇中一种或多种与水的混合溶剂。优选地,步骤(i)中所述极性溶剂1选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇或所述醇类溶剂的混合溶剂;以及甲醇/水混合溶剂、乙醇/水混合溶剂、丙醇/水混合溶剂、异丙醇/水混合溶剂、正丁醇/水混合溶剂以及异丙醇/水混合溶剂。
优选地,步骤(i)所述极性溶剂1为甲醇,乙醇,异丙醇,乙醇/水混合溶剂,异丙醇/水混合溶剂,甲醇/水混合溶剂。
更优选,步骤(i)所述极性溶剂1为甲醇。
优选地,步骤(i),化合物1粗品与极性溶剂1的质量比为1∶1~1∶10;优选为1∶4~1∶7;进一步优选为1∶4.5~1∶6.5。
优选地,步骤(i)所述的化合物1粗品的hplc纯度为小于95%,优选小于90%,优选小于85%;最优选80%~95%。
优选地,步骤(ii)所述加热所至的温度为30℃-100℃;优选40℃-80℃;更优选为50℃-70℃。
优选地,步骤(iii)所述的降温所至的温度为25℃-50℃,优选为35℃-45℃。
优选地,步骤(iii)所述的降温方式可以是自然降温或则通过仪器控制程序降温。
优选地,步骤(iii)所述的降温时间根据实际使用的设备热交换能力决定。
优选地,步骤(iii)所述加入的水与化合物1粗品的质量比为0∶1~3∶1;优选为0.2∶1~1∶1;更优选为0.4∶1~0.8∶1。
优选地,步骤(iii)中,所述的晶种的加入量为相当于化合物1粗品质量的
0~10%,优选的加入量为0.3%~1%。在一些实施例中,不加晶种也可以析晶得到高纯度的化合物1。
优选地,步骤(iii)中,加入水的方式为缓慢加入,优选缓慢滴加。防止加入过快导致的暴析,从而影响产品质量。
优选地,步骤(iv)所述的降温所至的温度为-20℃~20℃,优选为-5℃~10℃。
优选地,步骤(iv)所述的降温方式是自然降温,或者通过仪器控制程序降温。
优选地,步骤(v)中醇类溶剂选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇中的一种或多种。
优选地,步骤(v)中所述的分离为过滤。
优选地,步骤(v)所述的干燥为减压干燥,优选真空干燥。
优选地,步骤(v)所述的干燥温度为45℃~50℃下干燥,优选真空干燥,最优选减压真空干燥。
本发明的结晶方法选择醇类溶剂,或者醇/水混合溶剂作为结晶溶剂,且在降温析晶的过程中将温度降低至20℃以下,将获得纯度较高的bg-11,该工艺大大提高了大规模生产过程中中间体bg-11的纯度,提高了药品中间体的生产质量。
附图说明
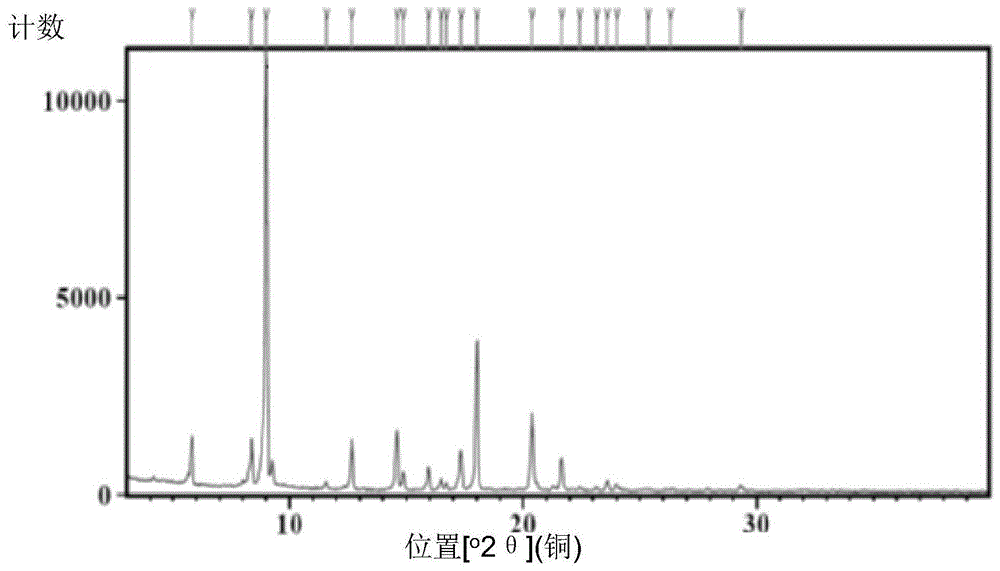
图1是实施例1中获得的化合物1(bg-11)的x射线衍射图。
图2是实施例1中获得的化合物1(bg-11)的dsc谱图。
具体实施方式
实施例1
含有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入67.5g甲醇,加热至55℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入8.25g水,降温至5℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到13.9g化合物1,纯度99.53%,该化合物的晶体x衍射图如图1所示,该化合物的dsc如图2所示。
实施例2
含有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入97.5g甲醇,加热至55℃(回流),搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入8.25g水,降温至5℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到13.13g化合物1,纯度99.46%。
实施例3
含有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入82.5g甲醇,加热至50℃,搅拌2h至澄清,降温至35℃,加入晶种,缓慢加入8.25g水,降温至5℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到13.96g化合物1,纯度99.48%。
实施例4
合有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入82.5g甲醇,加热至68℃(回流),搅拌2h至澄清,降温至43℃,加入晶种,缓慢加入8.25g水,降温至5℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到14.42g化合物1,纯度99.92%。
实施例5
含有16.5gbg-11粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入82.5g甲醇,加热至63℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入6.75g水,降温至5℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到15.07g化合物1,纯度100%。
实施例6
含有16.5gbg-11粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入82.5g甲醇,加热至63℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入9.75g水,降温至5℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到15.27g化合物1,纯度100%。
实施例7
合有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入82.5g甲醇,加热至63℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入8.25g水,降温至-3℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到15.47g化合物1,纯度99.82%。
实施例8
合有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入82.5g甲醇,加热至63℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入8.25g水,降温至13℃搅拌20h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到16.26g化合物1,纯度100%。
实施例9
合有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入67.5g乙醇,加热至63℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入9.75g水,降温至5℃搅拌20h,过滤,滤饼用乙醇/水混合溶液洗涤,45℃真空干燥12h,得到化合物1。
实施例10
含有16.5gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入60g异丙醇,加热至63℃,搅拌2h至澄清,降温至35℃,加入晶种,缓慢加入10.5g水,降温至5℃搅拌20h,过滤,滤饼用异丙醇/水混合溶液洗涤,45℃真空干燥12h,得到化合物1。
需要说明的是,本发明实施例所使用的晶种的获得方式与实施例的方式(除加入晶种外)一致,任何一个实施例描述的方法都可以用来制备晶种。首次小批量制备该晶型不需要晶种,后续制备和生产可以用之前批次的产品作为晶种。加晶种主要是为了更好的控制结晶过程,使产品平稳的析出,防止暴析。加晶种不是必须操作,不加晶种也可以结晶获得需要的晶体。
对比实例1
按照与实施例1相同的方式来获得化合物1的结晶物,不同之处在于,将实施例1中的醇类溶剂替换为乙腈,四氢呋喃,乙酸乙酯或在甲基叔丁基醚。然而,根据溶解度数据,室温下化合物1在乙腈,四氢呋喃,乙酸乙酯,甚至在甲基叔丁基醚中都有很好的溶解度,这些溶剂或者混合溶剂不合适作为化合物1的结晶溶剂。因此,使用这些溶剂无法获得化合物1的结晶物。
对比实例2
含有15gbg-11的粗品溶液通过蒸馏装置减压蒸馏除去溶剂,加入60g甲醇,加热至63℃,搅拌2h至澄清,降温至40℃,加入晶种,缓慢加入8.25g水,降温至25℃搅拌1h,过滤,滤饼用甲醇/水混合溶液洗涤,45℃真空干燥12h,得到13.41g化合物1。当降温至25℃时过滤,损失率近10%(w/w)。
由对比试验结果可知,室温下使用乙腈,四氢呋喃,乙酸乙酯、甲基叔丁基醚均不能实现结晶。化合物1(bg-11)在醇类或者醇类/水混合溶剂中的溶解度随温度变化而显著变化,在较高温度时有较高的溶解度,而降温后溶解度显著降低,是合适的化合物1结晶溶剂。为得到较高的回收率,优选降温到20℃以下,使产品充分析出。
上文中已经用一般性说明、具体实施方式和试验对本发明做了详尽的描述,在不偏离本发明精神的基础上所做的修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!