RGD序列肽修饰的六环哌嗪二酮,其制备,抗转移活性及应用的制作方法
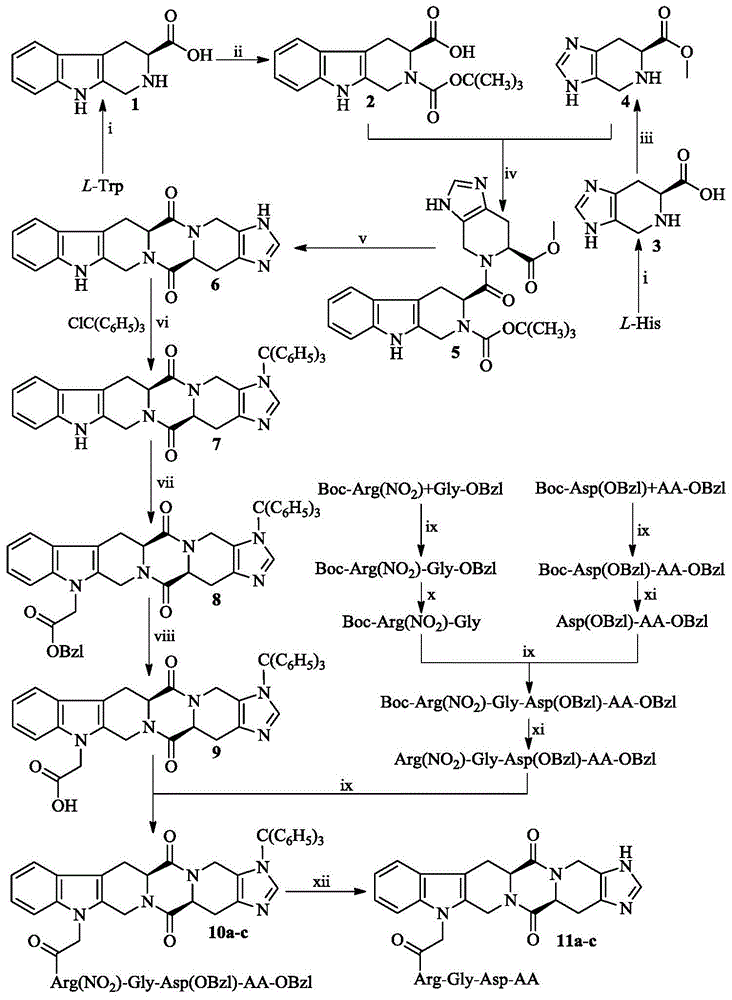
本发明涉及下9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑。涉及它们的制备方法,涉及它们在制备抗肿瘤转移药物中的应用。本发明属于生物医药领域。
背景技术:
肿瘤已经成为严重威胁人类健康的常见病。例如2015年新增的肿瘤患者大约有392.9万,其中有233.8万肿瘤患者死亡。平均每天有超过1万人被确诊为肿瘤。肿瘤转移,尤其是肿瘤向肺转移使得肿瘤死亡率逐年升高。肿瘤患者预后差的两个主要原因是:1)化疗药物的毒副作用严重;2)肿瘤转移尤其是向肺转移。目前,仍然没有能有效治疗肿瘤转移的药物用于临床。发明抗肿瘤转移药物是药物研究的前沿之一。
四氢-β-咔啉-3-羧酸是具有多种生物活性的药效团,环组氨酸也是具有多种生物活性的药效团。在一项相关发明中,发明人发现四氢-β-咔啉-3-羧酸与环组氨酸两个药效团融合形成的下式左的四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑是p-选择素抑制剂,具有抗肿瘤作用。进一步的研究使发明人认识到,在四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑的9位引入ch2co-arg-gly-asp-aa生成的下式右的9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(式中aa为ser残基,phe残基及val残基)还具有抗肿瘤转移作用。根据这种认识,发明人提出了本发明。
技术实现要素:
本发明的第一个内容是提供下式的9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(式中aa为ser残基,phe残基及val残基)。
本发明的第二个内容是提供制备9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(式中aa为ser残基,phe残基及val残基)的方法,该方法包括:
(1)l-trp在硫酸催化条件下与甲醛进行pictet-spengler反应,制备3s-1,2,3,4-四氢-β-咔啉-3-羧酸(1);
(2)在n,n-二甲基甲酰胺中1与(boc)2o反应,制备3s-2-叔丁氧羰基-1,2,3,4-四氢-β-咔啉-3-羧酸(2);
(3)l-his在硫酸催化条件下与甲醛进行pictet-spengler反应,制备6s-4,5,6,7-四氢-咪唑[4:5]并哌啶-6-羧酸(3);
(4)在甲醇和氯化亚砜中由3制备6s-4,5,6,7-四氢-咪唑[4:5]并哌啶-6-羧酸甲酯(4);
(5)以3-二乙氧基磷酰基-1,2,3-苯唑4(h)-酮作缩合剂,在无水四氢呋喃中2与4偶联制备2-叔丁氧羰基-四氢-β-咔啉-3-甲酰-哌啶[4:5]并咪唑-6-羧酸甲酯(5);
(6)脱除5的叔丁氧羰基保护基,溶于甲醇之后加n-甲基吗啉调ph为9制备四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶-[4:5]并咪唑(6);
(7)在二氯甲烷中6与三苯基氯甲烷反应,制备1-三苯甲基-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(7);
(8)在n,n-二甲基甲酰胺中7与溴乙酸苄酯反应,制备1-三苯甲基-9-乙酸苄酯-四氢-β-咔啉-[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(8);
(9)8脱苄基制备1-三苯甲基-9-乙酸-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(9);
(10)在n,n-二甲基甲酰胺中9与arg(no2)-gly-asp(obzl)-aa-obzl偶联,制备1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-aa-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10a-c,式中aa为ser残基,phe残基及val残基);
(11)10a-c脱保护制备9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(11a-c,式中aa为ser残基,phe残基及val残基)。
本发明的第三个内容是评价9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑对肿瘤细胞迁移和侵袭的抑制作用。。
本发明的第四个内容是评价9-(ch2co-arg-gly-asp-phe)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑抑制肿瘤向肺转移的作用。
附图说明
图1.9-(ch2co-arg-gly-asp-aa)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(式中aa为ser残基,phe残基及val残基)的合成路线。i)甲醛(37%),浓硫酸(98%),水,氨水溶液(25%);ii)n,n-二甲基甲酰胺,(boc)2o,三乙胺;iii)甲醇,氯化亚砜;iv)四氢呋喃,3-二乙氧基磷酰基-1,2,3-苯唑4(3h)-酮,三乙胺;v)氯化氢的乙酸乙酯溶液(4m),甲醇,n-甲基吗啉;vi)二氯甲烷,三苯基氯甲烷;vii)n,n-二甲基甲酰胺,氢化钠(60%),溴乙酸苄酯;viii)甲醇,钯碳,氢气;ix)n,n-二甲基甲酰胺,四氢呋喃,1-羟基苯并三唑,二环己基碳二亚胺,n-甲基吗啉;x)甲醇,氢氧化钠水溶液(2m);xi)氯化氢的乙酸乙酯溶液(4m);xii)三氟醋酸,三氟甲磺酸。
具体实施方式
为了进一步阐述本发明,下面给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1制备boc-arg(no2)-gly-obzl
0℃及搅拌下将1.595g(5.0mmol)boc-l-arg(no2)溶于20ml四氢呋喃,然后加0.675g(5.0mmol)1-羟基苯并三唑和1.236g(6.0mmol)二环己基碳二亚胺,反应0.5h。之后加1.854g(5.5mmol)tos·gly-obzl,最后反应液用n-甲基吗啉调ph为8。得到的溶液室温搅拌6h至tlc显示boc-l-arg(no2)完全消失。反应混合物过滤,滤液减压浓缩,得到的淡黄色油状物用60ml乙酸乙酯溶解,得到的溶液依次用饱和碳酸氢钠水溶液洗(20ml×3),饱和氯化钠水溶液洗(20ml×3),5%硫酸氢钾水溶液洗(20ml×3),饱和氯化钠水溶液洗(20ml×3),饱和碳酸氢钠水溶液洗(20ml×3),饱和氯化钠水溶液洗(20ml×3),乙酸乙酯层用无水硫酸钠干燥12h,过滤,滤液减压浓缩,得到2.120g(90%)标题化合物,为无色粉末,esi-ms(m/e):467[m+h]+。
实施例2制备boc-arg(no2)-gly
0℃及搅拌下将2.120g(5.0mmol)boc-arg(no2)-gly-obzl用15ml甲醇溶解,加氢氧化钠水溶液(2m)调ph为11,搅拌至tlc显示boc-arg(no2)-gly-obzl完全消失。反应溶液先用饱和硫酸氢钾水溶液调ph为中性,减压浓缩,再用饱和硫酸氢钾水溶液调ph为2,用乙酸乙酯萃取(50ml×3),合并乙酸乙酯层并用饱和氯化钠水溶液洗(30ml×3)。乙酸乙酯层用无水硫酸钠干燥12h,过滤,滤液减压浓缩得到1.542g(82%)标题化合物,为无色油状产物。esi-ms(m/e):375[m-h]-。
实施例3制备boc-asp(obzl)-ser-obzl
用实施例1的方法从1.615g(5.0mmol)boc-l-asp(obzl)和1.271g(5.5mmol)l-ser-obzl得到2.520g(92%)标题化合物,为无色油状产物。esi-ms(m/e):501[m+h]+。
实施例4制备asp(obzl)-ser-obzl
0℃及搅拌下将2.520g(5.0mmol)boc-asp(obzl)-ser-obzl用30ml氯化氢的乙酸乙酯溶液(4m)溶解,搅拌4h,tlc显示boc-asp(obzl)-ser-obzl完全消失。反应混合物减压浓缩,残留物用20ml无水乙酸乙酯溶液溶解。得到的溶液再减压浓缩。该操作重复3次。得到的固体用无水乙醚洗(10ml×3),得到1.930g(88%)标题化合物,为淡黄色油状物。esi-ms(m/e):401[m+h]+。
实施例5制备boc-arg(no2)-gly-asp(obzl)-ser-obzl
用实施例1的方法从1.542g(5.2mmol)boc-arg(no2)-gly和1.930g(5.0mmol)asp(obzl)-ser-obzl得到2.450g(69%)标题化合物,为无色粉末。esi-ms(m/e):757[m-h]-。
实施例6制备arg(no2)-gly-asp(obzl)-ser-obzl
用实施例4的方法从2.450g(3.2mmol)boc-arg(no2)-gly-asp(obzl)-ser-obzl得到1.820g(85%)标题化合物,为无色粉末。esi-ms(m/e):659[m+h]+。
实施例7制备boc-asp(obzl)-phe-obzl
用实施例1的方法从1.615g(5.0mmol)boc-l-asp(obzl)和1.604g(5.5mmol)l-phe-obzl得到1.993g(69%)标题化合物,为无色油状产物。esi-ms(m/e):561[m+h]+。
实施例8制备asp(obzl)-phe-obzl
用实施例4的方法从2.850g(5.0mmol)boc-asp(obzl)-phe-obzl得到2.230g(88%)标题化合物,为无色粉末。esi-ms(m/e):461[m+h]+。
实施例9制备boc-arg(no2)-gly-asp(obzl)-phe-obzl
用实施例1的方法从1.542g(5.2mmol)boc-arg(no2)-gly和2.340g(5.0mmol)asp(obzl)-phe-obzl得到2.924g(74%)标题化合物,为无色粉末。esi-ms(m/e):819[m+h]+。
实施例10制备arg(no2)-gly-asp(obzl)-phe-obzl
用实施例4的方法从2.924g(3.6mmol)boc-arg(no2)-gly-asp(obzl)-phe-obzl得到2.362g(92%)标题化合物,为无色粉末。esi-ms(m/e):719[m+h]+。
实施例11制备boc-asp(obzl)-val-obzl
用实施例1的方法从1.615g(5.0mmol)boc-l-asp(obzl)和1.342g(5.5mmol)l-val-obzl得到1.669g(65%)标题化合物,为无色油状产物。esi-ms(m/e):513[m+h]+。
实施例12制备asp(obzl)-val-obzl
用实施例4的方法从2.560g(5.0mmol)boc-asp(obzl)-val-obzl得到1.978g(96%)标题化合物,为无色粉末。esi-ms(m/e):413[m+h]+。
实施例13制备boc-arg(no2)-gly-asp(obzl)-val-obzl
用实施例1的方法从1.542g(5.2mmol)boc-arg(no2)-gly和2.060g(5.0mmol)asp(obzl)-val-obzl得到1.940g(62%)标题化合物,为无色粉末。esi-ms(m/e):771[m+h]+。
实施例14制备arg(no2)-gly-asp(obzl)-val-obzl
用实施例4的方法从1.940g(2.5mmol)boc-arg(no2)-gly-asp(obzl)-val-obzl得到1.589g(95%)标题化合物,为无色粉末。esi-ms(m/e):671[m+h]+。
实施例15制备3s-1,2,3,4-四氢-β-咔啉-3-羧酸(1)
0℃及搅拌下向200ml蒸馏水中加入0.1ml浓硫酸(98%),之后加入2.04g(10.0mmol)l-trp并搅拌至固体溶解,最后加入5ml甲醛水溶液(37%)并室温搅拌6h。tlc显示l-trp完全消失。0℃及搅拌下向反应混合物中加入氨水溶液(25%)调ph为7。反应混合物室温静置30min,过滤收集生成的沉淀,得到2.03g(93%)标题化合物,为淡黄色固体。esi-ms(m/e):217[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=12.141(s,1h),10.944(s,1h),7.436(s,1h),7.321(s,1h),7.032(d,2h),4.207(m,3h),2.822(m,2h)。
实施例16制备3s-2-叔丁氧羰基-1,2,3,4-四氢-β-咔啉-3-羧酸(2)
向0.972g(4.5mmol)3s-1,2,3,4-四氢-β-咔啉-3-羧酸(1)中加入10mln,n-二甲基甲酰胺,搅拌使固体悬浮。0℃及搅拌下先往该悬浮液中加入1.275g(1.3mmol)(boc)2o,再加入三乙胺调ph为10。得到的溶液室温搅拌至tlc显示化合物1完全消失。反应混合物减压浓缩,得到的淡黄色油状物用40ml乙酸乙酯溶解,得到的乙酸乙酯溶液先用5%硫酸氢钾水溶液洗(50ml×3)再用饱和氯化钠水溶液洗(50ml×3),乙酸乙酯层用无水硫酸钠干燥12h。过滤,滤液减压浓缩,得到的淡黄色固体在15ml二氯甲烷中超声,使固体均匀分散。过滤,得到1.106g(77%)标题化合物,为无色固体。esi-ms(m/e):315[m-h]-;1hnmr(300mhz,dmso-d6):δ/ppm=12.769(s,1h),10.868(d,1h),7.416(d,j=7.5hz,1h),7.283(m,1h),7.048(t,j=7.2hz,1h),6.966(t,j=7.2hz,1h),5.101(m,1h),4.716(t,j=12.9hz,1h),4.394(m,1h),3.297(d,j=16.2hz,1h),2.959(m,1h),1.461(d,j=9.9hz,9h)。
实施例17制备6s-4,5,6,7-四氢-咪唑[4:5]并哌啶-6-羧酸(3)
0℃及搅拌下向2.50g(16.1mmol)l-his与10ml蒸馏水的溶液中加入0.4ml浓硫酸(98%)使l-his逐渐溶解。向得到的溶液中加3ml甲醛水溶液(37%)并于60℃及加热6h,tlc显示l-his完全消失。反应混合物于0℃搅拌,之后加25%氨水溶液调ph为7。室温静置30min,过滤。收集的固体用水及丙酮各洗三次,得到2.649g(98%)标题化合物,为无色固体。esi-ms(m/e):168[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=12.016(s,1h),7.548(s,1h),5.040(d,j=13.8hz,1h),4.789(d,j=14.1hz,1h),4.277(m,1h),3.761(m,2h)。
实施例18制备6s-4,5,6,7-四氢-咪唑[4:5]并哌啶-6-羧酸甲酯(4)
0℃及搅拌下向120ml甲醇中滴加8ml氯化亚砜并搅拌40min。之后,加入5.01g(30mmol)6s-4,5,6,7-四氢-咪唑[4:5]并哌啶-6-羧酸(3)。反应混合物室温搅拌至tlc显示化合物3完全消失。反应混合物减压浓缩,残留物用50ml甲醇溶解。溶液减压浓缩,残留物再用50ml甲醇溶解。该操作重复3次。固体用无水乙醚洗(30ml×3),得到7.129g(93%)标题化合物,为无色粉末。esi-ms(m/e):182[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=12.667(s,1h),9.051(s,1h),4.710(m,1h),4.333(m,2h),3.819(s,3h),3.302(m,1h),3.154(m,1h)。
实施例19制备2-叔丁氧羰基-四氢-β-咔啉-3-甲酰哌啶[4:5]并咪唑-6-羧酸甲酯(5)
将2.212g(7.0mmol)3s-2-叔丁氧羰基-1,2,3,4-四氢-β-咔啉-3-羧酸(2),2.134g(8.4mmol)6s-4,5,6,7-四氢-咪唑[4:5]并哌啶-6-羧酸甲酯(4)以及2.512g(8.4mmol)3-二乙氧基磷酰基-1,2,3-苯唑4(3h)-酮用30ml四氢呋喃溶解。0℃与搅拌下向溶液中加3ml(21.0mmol)三乙胺,室温搅拌至tlc显示化合物4完全消失。滤去不溶物,滤液减压浓缩。得到的棕黄色糖浆状物用80ml二氯甲烷溶解,溶液用10%的碳酸钠水溶液洗(50ml×3)及饱和氯化钠水溶液洗(50ml×3)。二氯甲烷层用无水硫酸钠干燥12h,过滤。滤液减压浓缩,得到3.126g棕黄色固体。该固体经柱层析纯化,得到1.633g(52%)标题化合物,为无色粉末。esi-ms(m/e):502[m+na];1hnmr(300mhz,dmso-d6):δ/ppm=11.917(s,1h),10.823(s,1h),7.570(s,1h),7.436(m,1h),7.277(m,1h),7.068-6.911(m,2h),5.682-5.397(m,2h),4.896-4.435(m,4h),3.966(m,1h),3.633(d,j=5.7hz,1h),3.477(s,1h),3.396(s,1h),3.333(s,4h),3.186-2.813(m,5h),1.453-1.290(d,9h);13cnmr(125mhz,dmso-d6):δ/ppm=171.27,171.08,154.94,136.39,135.69,135.40,131.09,126.91,121.18,118.85,117.91,111.37,104.55,103.23,80.36,55.38,52.80,51.08,50.42,50.24,28.50,28.44,28.35,28.19,21.80。
实施例20制备四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(6)
0℃及搅拌下将1.633g(3.4mmol)2-叔丁氧羰基-四氢-β-咔啉-3-甲酰哌啶[4:5]并咪唑-6-羧酸甲酯(5)用25ml氯化氢的乙酸乙酯溶液(4m)溶解。得到的溶液在0℃搅拌4h,tlc显示化合物5完全消失。将反应混合物减压浓缩,残留物用20ml干燥的乙酸乙酯溶液溶解。得到的溶液再减压浓缩。该操作重复3次。得到的固体用无水乙醚洗(30ml×3),得到1.244g(95%)四氢-β-咔啉-3-甲酰哌啶[4:5]并咪唑-6-羧酸甲酯,为棕黄色粉末。esi-ms(m/e):380[m+h]+。
0℃及搅拌下将棕黄色四氢-β-咔啉-3-甲酰哌啶[4:5]并咪唑-6-羧酸甲酯粉末先用20ml甲醇溶解,再加n-甲基吗啉调ph为9,室温搅拌至tlc显示棕黄色粉末完全消失。反应混合物减压浓缩,得到的深棕色糖浆状物通过硅胶柱层析纯化,得到0.854g(72%)标题化合物,为淡黄色粉末。ft-esi-ms(m/e):348.1447[m+h]+;mp:210-211℃;1hnmr(300mhz,dmso-d6):δ/ppm=12.003(s,1h),11.017(s,1h),7.565(s,1h),7.414(d,j=7.8hz,1h),7.345(d,j=8.1hz,1h),7.072(t,j=7.5hz,1h),6.978(t,j=7.2hz,1h),5.427(d,j=16.5hz,1h),5.200(d,j=15.6hz,1h),4.493-4.455(m,2h),4.268(d,j=16.5hz,1h),4.063(m,1h),3.251(m,1h),3.075(m,1h),2.816(t,j=12.6hz,2h);13cnmr(125mhz,dmso-d6):δ/ppm=164.88,164.52,136.42,135.25,130.18,126.76,121.61,119.24,118.15,111.61,106.10,56.41,56.20,28.98,28.01。
实施例21制备1-三苯甲基-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(7)
0℃及搅拌下将0.500g(1.4mmol)四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(6)用15ml二氯甲烷溶解。之后,先加300μl(2.1mmol)三乙胺,再加0.400g(1.4mmol)三苯基氯甲烷。得到的溶液室温搅拌15h,tlc显示化合物6完全消失。加50ml冰水猝灭反应,水层用二氯甲烷萃取(20ml×3)。合并的二氯甲烷层用饱和氯化钠水溶液洗(20ml×3),用无水硫酸钠干燥12h,过滤,滤液减压浓缩,得到的棕黄色糖浆状物质经硅胶柱层析纯化,得到0.297g(35%)标题化合物,为无色粉末。esi-ms(m/e):612[m+na];1hnmr(300mhz,dmso-d6):δ/ppm=10.968(s,1h),7.442(s,10h),7.329(d,j=7.9hz,1h),7.247(s,1h),7.133-6.952(m,8h),5.373(d,j=16.5hz,1h),4.394(d,j=16.0hz,1h),4.309-4.158(m,3h),3.193(d,j=14.8hz,1h),3.013(d,j=14.3hz,1h),2.755(m,3h);13cnmr(125mhz,dmso-d6):δ/ppm=164.85,164.08,141.47,138.10,136.35,135.01,130.15,129.90,128.91,128.80,128.59,126.68,124.60,121.67,119.32,118.08,111.65,105.56,74.82,66.51,56.16,55.49,46.52,41.11,30.43,27.98。
实施例22制备1-三苯甲基-9-乙酸苄酯-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(8)
0℃及搅拌下将0.747g(1.3mmol)1-三苯甲基-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(7)用5mln,n-二甲基甲酰胺溶解。之后,加入0.101g(2.6mmol)氢化钠(60%)并搅拌5min。之后,加300μl(2.0mmol)溴乙酸苄酯。反应混合物室温搅拌5h,tlc显示化合物7完全消失。加50ml冰水猝灭反应,水层用乙酸乙酯萃取(30ml×3)。合并的乙酸乙酯层用饱和氯化钠水溶液洗(20ml×3),用无水硫酸钠干燥12h。过滤,滤液减压浓缩,得到的淡黄色油状物经硅胶柱层析纯化,得到0.386g(41%)标题化合物。为无色粉末。esi-ms(m/e):760[m+na];1hnmr(300mhz,dmso-d6):δ/ppm=7.498-7.365(m,16h),7.253(s,1h),7.139-7.027(m,8h),5.443(d,j=16.7hz,1h),5.188(s,4h),4.384(d,j=15.8hz,1h),4.232(m,2h),4.129(d,j=16.4hz,1h),2.232(d,j=14.9hz,1h),3.028(d,j=14.3hz,1h),2.893-2.694(m,4h);13cnmr(125mhz,dmso-d6):δ/ppm=170.81,169.37,164.34,164.32,141.35,137.87,137.28,136.10,135.40,131.40,130.01,128.92,128.90,128.75,128.62,128.38,126.58,123.93,121.94,119.97,118.50,109.94,106.93,75.02,66.89,60.23,56.28,56.07,44.93,39.17,31.23,27.80,21.24,14.56。
实施例23制备1-三苯甲基-9-乙酸-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(9)
将0.491g(0.6mmol)1-三苯甲基-9-乙酸苄酯-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(8)用20ml甲醇溶解。之后,加入5mgpd/c,用水泵抽气3min除瓶内空气,通氢气。该操作重复三次。反应混合物室温通氢气6h至tlc显示化合物8完全消失。反应混合物滤除pd/c,滤液减压浓缩,得到0.406g(94%)标题化合物,为无色粉末。esi-ms(m/e):646[m-h]-;1hnmr(300mhz,dmso-d6):δ/ppm=12.901(s,1h),7.459-7.012(m,20h),5.402(m,2h),4.982(s,2h),4.386(d,j=16.2hz,1h),4.224(m,2h),4.138(d,j=17.1hz,1h),3.225(d,j=16.2hz,1h),3.028(d,j=13.5hz,1h),2.767(m,2h);13cnmr(125mhz,dmso-d6):δ/ppm=170.83,164.78,164.32,144.25,141.31,137.84,137.25,135.29,131.38,130.01,129.51,128.93,128.81,128.76,128.74,128.35,126.71,126.47,123.97,121.83,119.78,118.41,109.82,106.55,75.07,56.37,56.29,56.24,56.04,55.91,44.94,39.14,31.15,27.80。
实施例24制备1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-ser-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10a)
用实施例1的方法从0.587g(0.8mmol)1-三苯甲基-9-乙酸-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(9)及0.629g(0.9mmol)arg(no2)-gly-asp(obzl)-ser-obzl得到0.528g(45%)标题化合物,为淡黄色粉末。esi-ms(m/e):1310[m+na]+;1hnmr(300mhz,dmso-d6):δ/ppm=8.573(d,j=7.5hz,2h),8.350(s,1h),8.272(d,j=7.5hz,2h),7.900(s,2h),7.444-7.341(m,24h),7.252(s,1h),7.136-6.988(m,9h),5.499(d,j=15.0hz,1h),5.094(m,5h),4.816(m,3h),4.381(m,3h),4.174(m,3h),3.724(m,4h),3.122(m,5h),2.755(m,5h),2.573(d,j=8.7hz,1h),1.724(s,1h),1.526(m,4h);13cnmr(125mhz,dmso-d6):δ/ppm=175.87,174.43,172.02,171.94,171.01,170.62,170.30,169.68,169.09,168.06,167.97,167.38,164.37,164.30,142.98,141.33,137.87,137.25,136.48,136.36,136.05,135.38,131.70,130.00,128.92,128.86,128.83,128.76,128.50,128.43,128.41,128.35,128.09,128.01,127.08,126.88,126.44,123.95,121.75,119.70,118.36,109.84,106.28,106.25,75.02,66.78,66.42,66.16,63.35,61.54,58.20,56.31,56.06,55.49,55.01,52.70,50.88,49.51,48.58,46.14,42.29,42.14,36.85,35.06,32.76,31.21,29.94,27.86,25.11。
实施例25制备1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-phe-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10b)
用实施例1的方法从0.587g(0.8mmol)1-三苯甲基-9-乙酸-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(9)及0.693g(0.9mmol)arg(no2)-gly-asp(obzl)-phe-obzl得到0.493g(40%)标题化合物,为淡黄色粉末。esi-ms(m/e):1370[m+na];1hnmr(300mhz,dmso-d6):δ/ppm=8.506(d,j=7.5hz,1h),8.413(d,j=7.2hz,1h),8.250(m,2h),7.856(s,2h),7.500(d,j=7.5hz,1h),7.423-6.975(m,42h),5.337(d,j=16.8hz,1h),5.167(d,j=15.6hz,1h),5.055(m,5h),4.791-4.711(m,3h),4.485(m,1h),4.314(m,3h),4.056(d,j=13.8hz,2h),3.711(m,2h),3.218(d,j=14.1hz,1h),3.112(m,2h),2.993(m,2h),2.683(m,1h),2.384(m,2h),2.201(d,j=13.8hz,1h),1.705(s,1h),1.705(s,1h),1.427(m,4h);13cnmr(125mhz,dmso-d6):δ/ppm=171.88,171.47,170.86,170.21,168.96,167.83,164.69,163.93,141.47,138.16,137.32,137.20,136.46,136.10,134.91,131.78,129.89,129.57,128.86,128.82,128.77,128.62,128.50,128.46,128.37,128.33,127.08,126.34,124.64,121.84,119.76,118.25,109.89,105.78,74.83,66.54,66.19,56.19,55.67,55.55,54.37,52.59,49.43,46.09,42.19,41.15,40.64,40.57,40.41,40.24,39.26,36.95,36.78,30.51,30.05,27.96。
实施例26制备1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-val-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10c)
用实施例1的方法从0.587g(0.8mmol)1-三苯甲基-9-乙酸-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(9)及0.650g(0.9mmol)arg(no2)-gly-asp(obzl)-val-obzl得到0.564g(47%)标题化合物,为淡黄色粉末。esi-ms(m/e):1322[m+na];1hnmr(300mhz,dmso-d6):δ/ppm=8.526(d,j=7.5hz,1h),8.285(m,2h),8.179(d,j=7.8hz,1h),7.871(s,1h),7.496(d,j=7.5hz,1h),7.369(m,20h),7.240(s,1h),7.094(m,8h),5.335(d,j=16.5hz,1h),5.111(m,5h),4.763(m,3h),4,315(m,3h),4.180(m,1h),4.061(m,2h),3.720(m,2h),3.385(d,j=6.9hz,1h),3.180(m,3h),2.703(m,1h),2.378(t,j=13.5hz,1h),2.195(d,j=15.0hz,1h),2.064(m,1h),1.704(m,1h),1.426(m,4h),0.842(d,j=6.3hz,6h);13cnmr(125mhz,dmso-d6):δ/ppm=171.93,171.88,171.53,171.09,170.24,169.18,169.16,167.97,167.88,164.71,164.38,164.29,163.93,141.45,141.32,138.16,137.88,137.25,137.20,136.46,136.26,135.35,134.88,131.76,131.70,129.99,129.89,128.92,128.87,128.83,128.77,128.63,128.57,128.51,128.45,128.37,128.23,127.99,126.45,126.33,124.64,123.96,121.84,121.75,119.70,118.35,109.83,106.25,105.77,75.03,74.84,66.42,66.18,60.24,58.12,56.31,56.19,56.06,55.54,52.71,49.54,46.12,42.20,40.64,36.65,31.20,30.24,29.95,27.86,19.36,18.57。
实施例27制备9-(ch2co-arg-gly-asp-ser)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(11a)
0℃及搅拌下将0.100g(0.07mmol)1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-ser-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10a)与1ml三氟乙酸和0.3ml三氟甲磺酸混合,搅拌0.5h至tlc显示10a完全消失。反应混合物在0℃及搅拌下用水泵抽气5min,加25ml冰冷的乙醚,溶液3000rpm离心5min,弃上清,再加25ml冰冷的乙醚,溶液3000rpm离心5min,弃上清,得到的墨绿色固体经c18纯化,得到15mg(22%)标题化合物,为黄色粉末。ft-esi-ms(m/e):819.3267[m-h]-;mp:172-173℃;1hnmr(300mhz,dmso-d6):δ/ppm=12.008(s,1h),10.006(s,1h),8.622(m,3h),7.564(s,1h),7.391(m,3h),7.129(m,1h),7.031(m,2h),5.568(d,j=16.2hz,1h),5.230(d,j=15.3hz,1h),4.941(m,2h),4.439(m,3h),4.263(m,2h),4.014(m,2h),3.877(m,1h),3.608(m,3h),3.454-3.056(m,7h),2.812(m,2h),1.976(s,1h),1.624(m,3h);13cnmr(125mhz,dmso-d6):δ/ppm=174.72,173.29,172.68,171.00,169.06,167.97,167.25,164.73,164.45,157.72,141.33,140.36,137.24,135.93,135.26,131.86,126.45,121.76,119.67,118.32,109.83,109.19,106.22,62.53,56.43,56.20,55.71,52.90,50.19,46.11,42.99,40.87,37.53,30.44,27.95,25.03;ir(cm-1):3247.09,3059.03,1647.83,1515.80,1463.31,1384.59,1326.16,1245.76,740.73,673.29。
实施例28制备9-(ch2co-arg-gly-asp-phe)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(11b)
用实施例27的方法从0.100g(0.07mmol)1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-phe-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10b)得到14mg(21%)标题化合物,为黄色粉末。ft-esi-ms(m/e):879.3617[m-h]-;mp:146-148℃;1hnmr(300mhz,dmso-d6):δ/ppm=12.053(s,1h),10.162(s,1h),8.822(d,j=7.2hz,1h),8.744(d,j=6.6hz,1h),8.466(d,j=12.9hz,1h),7.566(s,1h),7.402(m,3h),7.165(m,6h),7.009(m,3h),5.561(d,j=16.8hz,1h),5.225(d,j=15.0hz,1h),5.035(m,1h),4.891(m,2h),4.413(m,4h),4.222(m,2h),4.018(d,j=15.6hz,1h),3.908(m,1h),3.605(m,2h),2.918(m,9h),1.905(s,1h),1.558(m,3h);13cnmr(125mhz,dmso-d6):δ/ppm=173.73,170.55,169.07,167.90,167.17,164.73,164.56,164.45,157.77,141.33,140.37,138.72,137.24,135.94,135.28,131.88,129.86,128.69,128.32,126.37,121.75,119.68,109.19,106.22,56.43,56.14,55.05,52.74,50.72,46.12,42.88,40.82,40.65,40.58,39.32,39.14,37.89,37.21,30.38,27.94,24.63;ir(cm-1):3294.86,3053.21,1644.55,1514.94,1455.36,1381.50,1321.48,1241.62,1186.08,740.73,700.67。
实施例29制备9-(ch2co-arg-gly-asp-val)-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(11c)
用实施例27的方法从0.100g(0.07mmol)1-三苯甲基-9-[ch2co-arg(no2)-gly-asp(obzl)-val-obzl]-四氢-β-咔啉[3:4]并哌嗪-2,5-二酮并哌啶[4:5]并咪唑(10c)得到16mg(25%)标题化合物,为黄色粉末。ft-esi-ms(m/e):831.3595[m-h]-;mp:176-177℃;1hnmr(300mhz,dmso-d6):δ/ppm=12.094(s,1h),10.325(s,1h),9.021(d,j=6.3hz,1h),8.738(s,1h),8.638(m,1h),7.570(s,1h),7.427(m,1h),7.050(m,4h),5.552(d,j=14.7hz,1h),5.229(d,j=14.7hz,1h),4.971(m,2h),4.432(m,4h),4.241(m,1h),4.022(m,3h),3.532(d,j=19.2hz,1h),2.856(m,6h),2.308(d,j=16.8hz,1h),2.033(m,2h),1.621(m,3h),0.842(d,j=6.6hz,6h);13cnmr(125mhz,dmso-d6):δ/ppm=174.02,172.80,170.54,168.86,167.99,167.35,164.72,164.57,164.46,164.42,157.86,141.28,140.41,137.27,135.91,135.26,131.84,126.40,125.74,121.73,119.63,118.26,117.17,115.53,110.63,109.94,109.17,106.18,56.45,56.14,52.64,50.68,46.47,46.05,42.82,37.92,31.43,30.65,27.94,27.55,24.99,19.76,19.72,18.53,18.43;ir(cm-1):3249.90,2963.30,1651.33,1515.31,1463.21,1385.72,1326.66,1245.34,1187.49,740.73,667.67。
实施例30评价化合物11a-c抗肿瘤细胞迁移的活性
1)化合物6及11a-c用含0.5%二甲基亚砜的磷酸缓冲盐溶液配成浓度为500μm的溶液,经稀释加入96孔板,终浓度为20μm。阳性对照为rgds四肽,用含0.5%二甲基亚砜的磷酸缓冲盐溶液配成浓度为500μm的溶液,经过稀释加入96孔板,终浓度为20μm。空白对照为含0.5%二甲基亚砜的磷酸缓冲盐溶液。
2)青霉素,链霉素及8%多聚甲醛试液均购自solarbio公司。胎牛血清(fetalbovineserumaustraliasource,fbs)购自美国corning公司。二甲基亚砜及胰蛋白酶-edta消化液购自hyclone公司。用三蒸水将8.00g氯化钠,0.20g氯化钾,0.20g磷酸二氢钾及1.56g十二水合磷酸氢二钠搅拌溶解,转移至1000ml容量瓶中加水定容,高压蒸汽灭菌锅灭菌即得磷酸缓冲盐溶液。用三蒸水将60mg青霉素,100mg链霉素与一袋未开封的rpmi-1640培养基搅拌溶解,转移至1000ml容量瓶中加水定容即得rpmi-1640培养基,加入10%的胎牛血清并过0.22μm滤膜,瓶口处缠好密封膜,于4℃保存。结晶紫染液购自碧云天公司。
3)95d(高转移人非小细胞肺癌细胞)购自南京凯基公司。选取处于对数生长期的细胞,弃培养基加磷酸缓冲盐溶液洗(1ml×3)。之后,加入1ml胰蛋白酶-edta消化液,在含5%co2的孵箱中37℃孵育5min,显微镜下观察到80%细胞脱落。脱落的细胞加2ml完全培养基吹打使细胞混匀悬浮。3000rpm离心3min,弃上清,用无血清培养洗3次,再混匀悬浮。取少量细胞悬液计数,配制密度为4~6×105个/ml的细胞悬液。transwell上室加100μl细胞悬液,加25μl配制好的rgds或化合物6或化合物11a-c,轻轻晃动使混合混匀,设置空白对照。transwell下室加600μl完全培养基,置于孵箱中孵育8h。
吸去上室剩余液体,加30μl磷酸缓冲盐溶液,用棉签擦去上室细胞。该操作重复三次。吸去下室剩余液体,加600μl4%多聚甲醛试液固定细胞,时间为30min。取出小室,用蒸馏水清洗,吸去下室固定液,加600μl结晶紫染液细胞染色,时间为30min。取出小室,用蒸馏水洗净多余的结晶紫,用棉签擦净上室残余液体,晾干待拍照。
将晾干后的小室置于荧光倒置显微镜下拍照,每个小室选取9个不同的视野,固定角度与放大倍数,视野内的细胞应均匀分布,避免选取小室边缘处细胞视野,进行计数。用imagej软件统计照片中的细胞数,以均值±sd个表示,经t检验,p<0.05即有统计学差异。结果列入表1。可见,在20μm的浓度下化合物11a-c可有效地抑制95d细胞迁移。
表1化合物11a-c对95d细胞迁移的影响
a)与空白对照及化合物6比p<0.01,与rgds比p<0.05;b)与空白对照,化合物6及rgds比p<0.01;n=3.
实施例31评价化合物11a-c抑制肿瘤细胞侵袭的活性
1)化合物6及11a-c用含0.5%二甲基亚砜的磷酸缓冲盐溶液配成浓度为500μm的溶液,经稀释加入96孔板,终浓度为20μm。阳性对照为rgds四肽,用含0.5%二甲基亚砜的磷酸缓冲盐溶液配成浓度为500μm的溶液,经过稀释加入96孔板,终浓度为20μm。空白对照为含0.5%二甲基亚砜的磷酸缓冲盐溶液。
2)青霉素,链霉素及8%多聚甲醛试液均购自solarbio公司。胎牛血清(fetalbovineserumaustraliasource,fbs)购自美国corning公司。二甲基亚砜及胰蛋白酶-edta消化液购自hyclone公司。用三蒸水将8.00g氯化钠,0.20g氯化钾,0.20g磷酸二氢钾及1.56g十二水合磷酸氢二钠搅拌溶解,转移至1000ml容量瓶中加水定容,高压蒸汽灭菌锅灭菌即得磷酸缓冲盐溶液。用三蒸水将60mg青霉素,100mg链霉素与一袋未开封的rpmi-1640培养基搅拌溶解,转移至1000ml容量瓶中加水定容即得rpmi-1640培养基,加入10%的胎牛血清并过0.22μm滤膜,瓶口处缠好密封膜,于4℃保存。结晶紫染液购自碧云天公司。
3)将matrigel基质胶从-20℃冰箱中取出,于4℃冰箱中过夜使变为液态。用无血清培养基将matrigel基质胶稀释10倍,加入transwell的上室,每室加100μl,于5%co2的孵箱中37℃孵育5h。吸除上室中的残余液体,沿内壁加50μl无血清的培养基,置孵箱中孵育30min,吸去上室残余液体。
选取处于对数生长期的细胞,弃培养基,加磷酸缓冲盐溶液洗(1ml×3)。之后,加入1ml胰蛋白酶-edta消化液,置孵箱中孵育5min,显微镜下观察到80%细胞脱落。将脱落的细胞在2ml完全培养基中吹打混匀,3000rpm离心3min,弃上清,用无血清培养洗3次,再混匀悬浮。取少量细胞悬液计数,配制密度为4~6×105个/ml的细胞悬液。每个transwell上室加100μl细胞悬液,加25μl配制好的rgds或化合物6或化合物11a-c,轻轻晃动使混合混匀,设置空白对照。transwell下室加600μl完全培养基,置孵箱中孵育12h。
吸去上室剩余液体,加30μl磷酸缓冲盐溶液,用棉签擦去上室细胞。该操作重复三次。吸去下室剩余液体,加600μl4%多聚甲醛试液固定细胞,时间为30min。取出小室,用蒸馏水清洗,吸去下室固定液,加600μl结晶紫染液细胞染色,时间为30min。取出小室,用蒸馏水洗净多余的结晶紫,用棉签擦净上室残余液体,晾干待拍照。
将晾干后的小室置于荧光倒置显微镜下拍照,每个小室选取9个不同的视野,固定角度与放大倍数,视野内的细胞应均匀分布,避免选取小室边缘处细胞视野,进行计数。用imagej软件统计照片中的细胞数,以均值±sd个表示,经t检验,p<0.05即有统计学差异。结果列入表2。可见,在20μm的浓度下化合物11a-c可有效地抑制95d细胞侵袭。
表2化合物11a-c对95d细胞侵袭的影响
a)与空白对照比p<0.01,与化合物6及rgds比p>0.05;b)与空白对照,化合物6及rgds比p<0.01;n=3.
实施例32评价化合物11a-c抑制肿瘤肺转移的活性
1)阳性对照为rgds四肽,腹腔注射剂量为20μmol/kg/天,连续给药10天;空白对照为生理盐水,口服剂量为0.1ml/10g/天,连续给药10天;化合物6的口服剂量为0.1μmol/kg/天,连续给药10天;化合物11a-c的口服剂量为0.01μmol/kg/天,连续给药10天。
2)lewis小鼠肺癌细胞(llc)购自atcc。用含10%胎牛血清的dmem完全培养基对llc传代培养,得到处于对数生长期的细胞。弃培养基,残余细胞先用磷酸缓冲盐溶液洗(1ml×3),再用1ml胰蛋白酶-edta消化液消化,置5%co2的孵箱中37℃孵育1min,显微镜观察80%细胞脱落。脱落的细胞加2ml含血清的培养基吹打使悬浮均匀,3000rpm离心3min。弃上清,加生理盐水吹打,使细胞重悬浮均匀。取少量细胞悬液计数,配制细胞密度为2×107个/ml的细胞悬液,作接种瘤液。
体重20±2g的清洁级雄性c57bl/6小鼠,购自北京维通利华动物实验技术有限公司。将瘤液接种于小鼠右侧腋下,每只小鼠接种0.2ml。每日观察,~20天可明显见到小鼠右侧腋下长出直径约为~2.0cm的实体瘤,用作瘤源。取生长状况良好的瘤源鼠,经乙醚麻醉,颈椎脱臼处死,用75%乙醇溶液浸泡消毒,钝性分离瘤源鼠的实体瘤,置无菌表面皿中在4℃预冷的生理盐水中剔除坏死部分与浮血。剪碎剩余的状况良好的瘤组织,按体积比1/1加生理盐水,转移至玻璃组织匀浆器内研磨制备肿瘤匀浆,将肿瘤匀浆用200目的尼龙筛网过滤两次,得到单细胞悬液,取少部分悬液加生理盐水稀释计数,配制密度为2×107个/ml的细胞悬液,用台盼蓝染色法确保活细胞数大于95%。
将细胞悬液接种于雄性c57bl/6小鼠右侧腋下,每只小鼠接种0.2ml。每日观察,~15天可见到小鼠右侧腋下长出直径为4~5mm的实体瘤,按照肿瘤体积将小鼠随机分组,使各组小鼠的肿瘤体积均匀分布,每组12只小鼠。之后,各组小鼠连续给药10天,第十一天记录各组小鼠的体重,乙醚麻醉后颈椎脱臼处死,钝性分离小鼠肺部,为准确统计肺上转移的瘤结数,小鼠的肺要求完整无损。瘤结数以均值±sd个表示,经t检验p<0.05即有统计学差异。结果列入表3。肺上转移的瘤结数表明,在0.01μmol/kg/天的口服剂量下化合物11a-c有效地抑制肿瘤肺转移。其中化合物11c疗效显著强于腹腔注射剂量为20μmol/kg/天的rgds及口服剂量为0.1μmol/kg/天的化合物6。可见,本发明有突出的技术效果。
表3化合物11a-c抑制肿瘤肺转移活性
a)与生理盐水比p<0.01,与rgds比p<0.05,与化合物6比p>0.05;b)与生理盐水,化合物6及rgds比p<0.01;n=12。
- 还没有人留言评论。精彩留言会获得点赞!