一种苯并噻唑酯类衍生物的合成方法与流程
本发明属于有机合成领域,具体地,涉及一种苯并噻唑酯类衍生物的合成方法。
背景技术:
苯并噻唑类化合物广泛存在于天然产品中,因其具有良好的生物与生理活性,而被广泛的应用于农药、医药行业等方面。工业上,是制备荧光染料、荧光增白剂的原料,医药上,用于肿瘤、类风湿性关节炎等疾病的治疗以及多种菌株和癌细胞系的检测。因其在生产、医药领域重要的应用价值,对其合成方法的研究尤为重要。
文献中报道过的合成苯并噻唑类化合物的方法主要有以下几种:
(一)2013年,wu课题组报道了以pd(oac)2催化芳烃邻位c(sp2)-h酰基化的实验方案。(二)同年,ding课题组发展了钯催化芳烃的邻位酰基化的反应。(三)2014年,kuang课题组发展了钯催化1,2,3-三唑类化合物邻位酰氧基化的反应。(四)2015年,patel课题组通过催化剂pd(oac)2、氧化剂硝酸铈铵(can)实现了苯并噻唑酯类衍生物邻位发生酯基化、芳氧基化的反应。(五)2017年,chakraborti使用nhpi为添加剂,以pd(ii)-催化活化c(sp2)-h键,实现了杂环骨架与芳基甲烷交叉脱氢偶联(cdc)的芳基化过程。
综上所述,综上所述,现有技术合成苯并噻唑酯类衍生物化合物的方法很多,但是这些方法有些需要经过复杂的合成步骤且副反应较多和产率比较低;有些使用高毒性的底物或有机溶剂易造成环境污染;有些使用贵金属钯等为催化剂,成本较为昂贵,不适用于工业生产等缺点。因此,提供一种合成苯并噻唑酯类衍生物的新型的绿色合成方法很有必要。
技术实现要素:
为解决现有技术中存在的不存,本发明提供了一种合成苯并噻唑酯类衍生物的合成方法,以银盐为氧化剂,廉价的铜催化剂诸如(醋酸铜、硫酸铜、溴化亚铜、氧化亚铜和碘化亚铜中的一种或多种)催化苯并噻唑衍生物与有机羧酸在有机溶剂中反应,即可得到苯并噻唑酯类衍生物,该方法不用使用高毒性的底物或有机溶剂,也不需要贵金属钯等贵催化剂,本发明中的原料易得,合成步骤简单,副反应少,目标产物产率高,推广价值高。
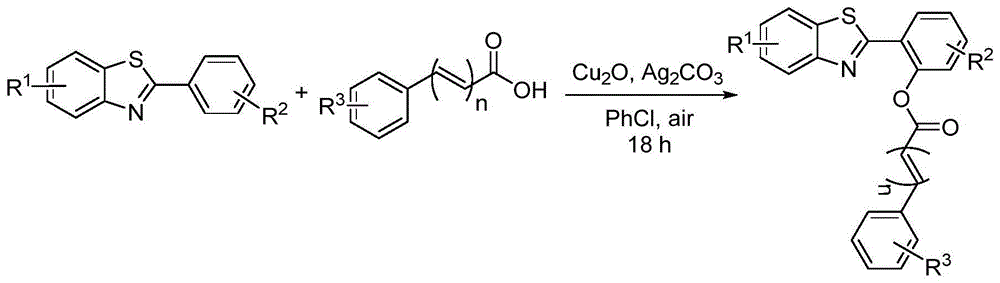
为了实现上述目的,本发明提供了一种苯并噻唑酯类衍生物的合成方法,包括:在混合条件下,将式ⅰ所示的苯并噻唑衍生物、有机羧酸、铜催化剂和银盐于有机溶剂中加热反应,得到如式ⅱ所示的苯并噻唑酯类衍生物,式ⅱ中x为所述有机羧酸中的酯基;
通过上述技术方案,本发明克服了现有技术中合成苯并噻唑酯类衍生物存在的诸多缺陷,提供了一种合成苯并噻唑酯类衍生物的合成方法,以银盐为氧化剂,廉价的铜催化剂诸如(醋酸铜、硫酸铜、溴化亚铜、氧化亚铜和碘化亚铜中的一种或多种)催化苯并噻唑衍生物与有机羧酸在有机溶剂中反应,即可得到苯并噻唑酯类衍生物。与现有技术相比,本发明有以下优点:(1)在空气气氛中进行,成本低;(2)没有使用钯、钌等贵金属,而是利用氧化亚铜等作催化剂,节约成本;(3)反应原料低廉且易得;(4)该合成方法效率高,使用范围广,适合多种底物反应。该方法在空气气氛中进行,成本低,不用使用高毒性的底物或有机溶剂,也不需要贵金属钯等贵催化剂,本发明中的原料易得,合成步骤简单,副反应少,目标产物产率高,推广价值高。
本发明的其他特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
图1为2-邻甲基苯基苯并噻唑与芳香酸的反应方程式;
图2a为制备的2-苯并噻唑-3-甲基苯基苯甲酸酯的1hnmr。
图2b为制备的2-苯并噻唑-3-甲基苯基苯甲酸酯的13cnmr。
图3a为制备的2-苯并噻唑-3-甲基苯基-4-甲氧基苯甲酸酯的1hnmr。
图3b为制备的2-苯并噻唑-3-甲基苯基-4-甲氧基苯甲酸酯的13cnmr。
图4a为制备的2-苯并噻唑-3-甲基苯基-4-氯苯甲酸酯的1hnmr。
图4b为制备的2-苯并噻唑-3-甲基苯基-4-氯苯甲酸酯的13cnmr。
图5a为制备的2-苯并噻唑-3-甲基苯基-2-噻吩羧酸酯的1hnmr。
图5b为制备的2-苯并噻唑-3-甲基苯基-2-噻吩羧酸酯的13cnmr。
图6a为制备的2-苯并噻唑-3-甲基苯基丙酸酯的1hnmr。
图6b为制备的2-苯并噻唑-3-甲基苯基丙酸酯的13cnmr。
图7a为制备的2-苯并噻唑-4-甲基苯基苯甲酸酯的1hnmr。
图7b为制备的2-苯并噻唑-4-甲基苯基苯甲酸酯的13cnmr。
图8a为制备的2-(6′-甲氧基苯并噻唑)-3-甲基苯基苯甲酸酯的1hnmr。
图8b为制备的2-(6′-甲氧基苯并噻唑)-3-甲基苯基苯甲酸酯的13cnmr。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明提供了一种苯并噻唑酯类衍生物的合成方法,其特征在于,包括:在混合条件下,将式ⅰ所示的苯并噻唑衍生物、有机羧酸、铜催化剂和银盐于有机溶剂中加热反应,得到如式ⅱ所示的苯并噻唑酯类衍生物,式ⅱ中x为所述有机羧酸中的酯基;
通过上述技术方案,本发明克服了现有技术中合成苯并噻唑酯类衍生物存在的诸多缺陷,提供了一种合成苯并噻唑酯类衍生物的合成方法,以银盐为氧化剂,廉价的铜催化剂诸如(醋酸铜、硫酸铜、溴化亚铜、氧化亚铜和碘化亚铜中的一种或多种)催化苯并噻唑衍生物与有机羧酸在有机溶剂中反应,即可得到苯并噻唑酯类衍生物。与现有技术相比,本发明有以下优点:(1)在空气气氛中进行,成本低;(2)没有使用钯、钌等贵金属,而是利用氧化亚铜等作催化剂,节约成本;(3)反应原料低廉且易得;(4)该合成方法效率高,使用范围广,适合多种底物反应。该方法在空气气氛中进行,成本低,不用使用高毒性的底物或有机溶剂,也不需要贵金属钯等贵催化剂,本发明中的原料易得,合成步骤简单,副反应少,目标产物产率高,推广价值高。
在上述技术方案中,有机羧酸可以有多种选择,苯并噻唑衍生物和不同的羧酸上述技术方案下反应,可以得到不同的酯基取代的苯并噻唑酯类衍生物。为了提高目标产物的产率,在本发明一种优选的实施方式中,优选地,所述有机羧酸为噻吩-2-甲酸、丙酸、2-苯基丁酸或如式ⅲ所示的芳香酸;
在上述技术方案中,各原料的用量可在较宽范围内进行选择,在本发明一种优选的实施方式中,为了提高目标产物的产率,减少副反应,并利于后续产物的纯化,优选地,式ⅰ所示的苯并噻唑衍生物、有机羧酸、铜催化剂和银盐的摩尔比为1:1.5-2.2:0.05-0.2:1-2。
在上述技术方案中,加热反应的条件可在较宽范围内调整,在本发明一种优选的实施方式中,为了提高目标产物的产率,减少副反应,并利于后续产物的纯化,优选地,加热反应的温度为130℃-140℃。更进一步地,反应过程不需要隔绝空气,可以在接触空气的情况下进行。
在上述技术方案中,加热反应的条件可在较宽范围内调整,在本发明一种优选的实施方式中,为了提高目标产物的产率,减少副反应,并利于后续产物的纯化,优选地,反应时间为18-24h。
在上述技术方案中,铜催化剂可以有多种选择,在本发明一种优选的实施方式中,为了提高目标产物的产率,减少副反应,优选地,铜催化剂为醋酸铜、硫酸铜、溴化亚铜、氧化亚铜和碘化亚铜中的一种或多种。选用不同的铜催化剂,得到产物的产率差别不大,均在10%以内,在后文的实施例中,以氧化亚铜进行说明。
在上述技术方案中,银盐可以有多种选择,在本发明一种优选的实施方式中,为了提高目标产物的产率,减少副反应,优选地,银盐为碳酸银、醋酸银和氧化银中的一种或多种。选用不同的银盐,得到产物的产率差别不大,均在10%以内,在后文的实施例中,以碳酸银进行说明。
在上述技术方案中,有机溶剂可以有多种选择,在本发明一种优选的实施方式中,为了提高目标产物的产率,减少副反应并利于产物的纯化,优选地,所述有机溶剂选自氯苯和、或对二甲苯中的一种或多种。在后文的实施例中,以氯苯进行说明。
在上述技术方案中,为了得到目标产物,在本发明一种优选的实施方式中,还包括对加热反应后得到的混合物分离纯化的步骤。
对于目标产物的纯化过程,可以采用常规的方式进行,为了简化纯化步骤,并得到较高产率的目标产物,在本发明一种优选的实施方式中,所述分离纯化的步骤包括:将对加热反应后得到的混合物用乙酸乙酯萃取后,用无水硫酸钠干燥,然后减压浓缩,得粗产物;以体积比为石油醚:乙酸乙酯=10:1的混合溶剂为展开剂,通过柱层析分离,得到苯并噻唑酯类衍生物。
以下将通过实施例对本发明进行详细描述。
实施例1
2-苯并噻唑-3-甲基苯基苯甲酸酯的合成,包括以下步骤:
a、取0.2mmol邻甲基苯基苯并噻唑,0.4mmol苯甲酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得白色固体。结果如图2a和图2b所示,证实为目标产物,经计算,产率83%。
1hnmr(500mhz,cdcl3):δ8.06(d,j=8.0hz,1h),7.94(d,j=7.5hz,2h),7.85(d,j=8.0hz,1h),7.52-7.45(m,3h),7.39-7.32(m,3h),7.27(d,j=7.5hz,1h),7.22(d,j=8.0hz,1h),2.38(s,3h).13cnmr(125mhz,cdcl3):δ165.1,162.8,153.2,149.4,139.8,136.1,133.6,130.6,130.2,129.1,128.5,128.2,127.0,126.0,125.3,123.5,121.5,120.1,20.4.hrms(esi):calcdforc21h16no2s[m+h]+364.0902,found346.0910.
个人猜想原理为:首先,cu2o被氧化,生成的活性催化剂与2-苯基苯并噻唑形成环金属化中间体,之后被苯甲酸自由基氧化成高活性的cu(ⅲ)中间体,最后,经过还原消除过程,释放出产物。反应过程如图1所示。
实施例2
2-苯并噻唑-3-甲基苯基-4-甲氧基苯甲酸酯的合成,包括以下步骤:
a、取0.2mmol邻甲基苯基苯并噻唑,0.4mmol4-甲氧基苯甲酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得无色液体。结果如图3a和图3b所示,证实为目标产物,经计算,产率73%
1hnmr(500mhz,cdcl3):δ7.97(d,j=8.0hz,1h),7.81-7.79(m,2h),7.73(d,j=8.0hz,1h),7.37-7.32(m,2h),7.27-7.24(m,1h),7.15-7.09(m,2h),6.71-6.69(m,2h),3.67(s,3h),2.27(s,3h).13cnmr(100mhz,cdcl3):δ164.5,163.5,162.6,152.9,149.1,139.4,135.8,132.0,130.3,127.8,126.7,125.7,124.9,123.2,121.2,121.0,113.4,55.1,20.1.hrms(esi):calcdforc22h18no3s[m+h]+376.1007,found376.1003。
实施例3
2-苯并噻唑-3-甲基苯基-4-氯苯甲酸酯的合成,包括以下步骤:
a、取0.2mmol邻甲基苯基苯并噻唑,0.4mmol4-氯苯甲酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得黄色液体。结果如图4a和图4b所示,证实为目标产物,经计算,产率83%
1hnmr(500mhz,cdcl3):δ7.96(d,j=8.0hz,1h),7.77-7.74(m,3h),7.39-7.34(m,2h),7.30-7.28(m,1h),7.21-7.16(m,3h),7.12(d,j=8.5hz,1h),2.28(s,3h).13cnmr(100mhz,cdcl3):δ164.2,162.6,153.2,149.2,140.1,139.9,136.0,131.6,130.7,128.9,128.4,127.5,127.0,126.2,125.4,123.6,121.6,120.5,20.4.hrms(esi):calcdforc21h15clno2s[m+h]+380.0512,found380.0507.
实施例4
2-苯并噻唑-3-甲基苯基-2-噻吩羧酸酯的合成,包括以下步骤:
a、取0.2mmol邻甲基苯基苯并噻唑,0.4mmol噻吩-2-甲酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得黄色液体。结果如图5a和图5b所示,证实为目标产物,经计算,产率41%
1hnmr(400mhz,cdcl3):δ8.10(d,j=8.0hz,1h),7.90(d,j=7.6hz,1h),7.77(dd,j=3.6,1.2hz,1h),7.54-7.46(m,3h),7.43-7.39(m,1h),7.30-7.28(m,2h),7.03(dd,j=5.2,4.0hz,1h),2.40(s,3h).13cnmr(100mhz,cdcl3):δ162.7,160.4,153.2,148.9,139.9,136.2,134.9,133.8,132.3,130.7,128.4,128.0,127.0,126.1,125.3,123.6,121.6,120.5,20.4.hrms(esi):calcdforc19h14no2s2[m+h]+352.0466,found352.0467.
实施例5
2-苯并噻唑-3-甲基苯基丙酸酯的合成,包括以下步骤:
a、取0.2mmol邻甲基苯基苯并噻唑,0.4mmol丙酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得黄色液体。结果如图6a和图6b所示,证实为目标产物,经计算,产率53%
1hnmr(400mhz,cdcl3):δ8.13(d,j=8.0hz,1h),7.94(d,j=8.0hz,1h),7.55-7.51(m,1h),7.46-7.39(m,2h),7.06(d,j=8.0hz,1h),7.22(d,j=7.6hz,1h),2.36-2.30(m,5h),1.00(t,j=7.6hz,3h).13cnmr(100mhz,cdcl3):δ173.0,163.0,153.4,149.3,139.7,136.3,130.7,128.2,127.1,126.3,125.5,123.7,121.7,120.6,27.7,20.4,9.1.hrms(apci):calcdforc17h16no2s[m+h]+298.0901,found298.0898.
实施例6
2-苯并噻唑-4-甲基苯基苯甲酸酯的合成,包括以下步骤:
a、取0.2mmol间甲基苯基苯并噻唑,0.4mmol苯甲酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得白色固体。结果如图7a和图7b所示,证实为目标产物,经计算,产率45%
1hnmr(400mhz,cdcl3):δ8.32(dd,j=8.4,1.2hz,2h),8.23(s,1h),7.95(d,j=8.0hz,1h),7.81(d,j=8.0hz,1h),7.23-7.68(m,1h),7.59-7.55(m,2h),7.47-7.42(m,1h),7.38-7.32(m,2h),7.22(d,j=8.0hz,1h),2.49(s,3h).
13cnmr(100mhz,cdcl3):δ165.5,162.8,152.9,146.7,136.6,135.6,134.0,132.5,130.9,130.6,129.7,128.9,126.4,126.1,125.4,123.8,123.4,121.6,21.0.hrms(apci):calcdforc21h16no2s[m+h]+346.0902,found346.0901.
实施例7
2-(6′-甲氧基苯并噻唑)-3-甲基苯基苯甲酸酯的合成,包括以下步骤:
a、取0.2mmol2-(2-甲基苯基)-6-甲氧基苯并噻唑,0.4mmol苯甲酸置于反应管中,再然后依次加入0.02mmolcu2o、0.4mmolag2co3、3mlphcl,在140℃下搅拌反应18h。
b、将产物用乙酸乙酯进行萃取,无水硫酸钠干燥,减压浓缩,得粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=10:1)纯化得无色液体。结果如图8a和图8b所示,证实为目标产物,经计算,产率88%
1hnmr(400mhz,cdcl3):δ7.97-7.91(m,3h),7.51(t,j=7.6hz,1h),7.44(t,j=8.0hz,1h),7.35(t,j=8.0hz,2h),7.28-7.25(m,2h),7.19(d,j=8.0hz,1h),7.06(dd,j=9.2,2.8hz,1h),3.84(s,3h),2.38(s,3h).13cnmr(100mhz,cdcl3):δ165.3,160.1,157.8,149.5,147.9,140.0,137.6,133.7,130.6,130.4,129.2,128.6,128.3,127.1,124.1,120.6,115.7,103.8,55.9,20.6.hrms(apci):calcdforc22h18no3s[m+h]+376.1007,found376.1014
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!