一类新型光敏剂的制备方法及应用与流程
本发明涉及光动力治疗技术领域,具体涉及一类新型光敏剂、制备方法及其应用。
背景技术:
光动力治疗(pdt)是以非手术的形式治疗癌症的一种手段。1975年,dougherty等人首次对其进行论证,并进行了广泛的研究。目前,pdt已成为一种癌症治疗的特殊方式。光动力疗法在光照条件下利用光敏剂吸收光子,达到激发态,通过隙间穿越,将能量传递给三线态氧,并将三线态氧转化为具有细胞毒性的单线态氧(1o2),进而不可逆地杀伤癌细胞,达到治疗癌症的目的。相比于化疗和放疗,光动力疗法无创伤、无毒副作用、可重复治疗。
近年来,光动力疗法虽然已经引起人们的广泛关注,但是在实际应用中,治疗效果并不理想,因此设计一种性能优良的光敏剂对于光动力疗法而言尤为重要。
技术实现要素:
本发明提供了一类新型光敏剂的制备方法及其应用,解决了现有光敏剂吸收范围窄、可见光范围内治疗效果差、使用光源功率过大、无法引入官能团的技术问题。
本发明提供了一类新型光敏剂,具有如式(i)所示的结构式:
式(i)和(ii)中q为o或s或se。
式(i)和(ii)中r1为h或
式(ii)中x为h或br或i。
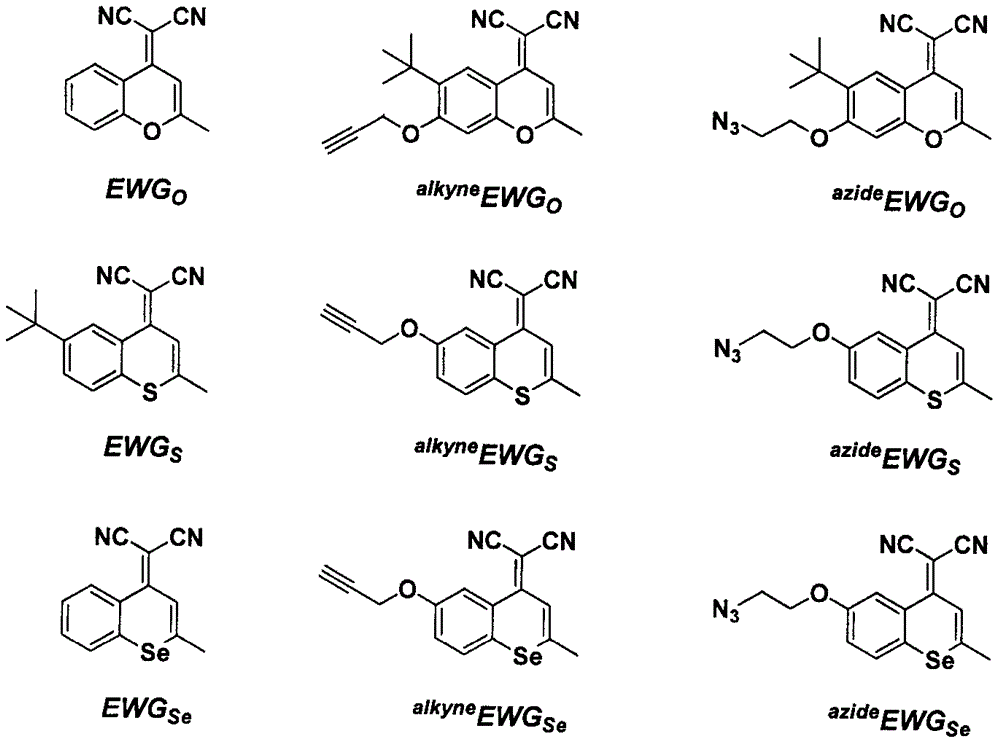
本发明以式(i)中的结构式为母核,创造性地进行改造母核,将中心原子o更换为s和se,并在r1和r2位置预留官能团,得到其衍生物,具体结构见附图1,合成步骤见附图3。
本发明提供了所述光敏剂的制备方法,将式(i)中的母核分别与对羟基苯甲醛、3,5-二溴-4-羟基苯甲醛和3,5-二碘-4-羟基苯甲醛进行knoevenagel反应,得到一类新型光敏剂,具体结构见附图2,具体合成步骤见附图4。
作为优选,knoevenagel反应的溶剂为乙腈,温度为85℃。
作为优选,knoevenagel反应中的哌啶和乙酸的体积比为2∶1。
作为优选,引入卤(br、i)原子,一方面可有效降低光敏剂酚羟基的pka,使其更加适应生理ph值(se代光敏剂为例,见附图5),另一方面促进光敏剂隙间穿越,提高光敏性。
作为优选,相同浓度下该类光敏剂与传统的光敏剂(mb)相比较,在可见光区有较大的吸收面积,见附图6。
作为优选,中心原子q(o、s、se)所对应的光敏剂,其波长依次红移(x为br,o<s<se;x为i,o<s<se,见附图7),该类光敏剂具有较大的斯托克位移,有效避免了光敏剂的自吸收,减少能量损失。
此外,本发明还提供了一种光动力测试方法,将光敏剂很好地分散在pbs溶液中,测试方法如下:
1)dma(60μm)、f127(0.3%)和光敏剂(2.5μm)溶于dmf,混合均匀,分散到pbs溶液中。
2)氙灯光源(490-700nm),5mw/cm2,每光照一分钟,使用紫外分光光度计依次扫描,扫描范围350-800nm,共计光照10min。
本发明实施例中引入硒元素的光敏剂,可以发挥重原子效应,有效促进隙间穿越,并且硒为人体重要的组成元素,因此,硒元素的引入可以降低光敏剂的生物毒性,并进一步提高光动力治疗的效果。
附图说明
图1所述光敏剂重要中间体的结构式。
图2所述光敏剂的结构式。
图3所述光敏剂主要中间体的合成路线。
图4所述光敏剂的合成路线。
图5以se代光敏剂为例,在不同的ph条件下的紫外吸收。
图6相同浓度下部分光敏剂与传统光敏剂(mb)的紫外吸收。
图7所述光敏剂的紫外吸收。
图8所述光敏剂重要中间体及光敏剂结构通式。
具体实施方式
下面结合附图和具体实施例对本发明做出进一步的解释和说明,基于本发明中的实施例,本领域的技术人员,在没有做出创造性的劳动成果下所获得的其他实施例,都属于本发明的保护范围。
实施例一:合成ewgo、alkyneewgo、azideewgo,合成路线见附图3
1)邻羟基苯乙酮(10g,73.45mmol)溶于乙酸乙酯中,加入钠(7g,0.3mol),然后85℃搅拌4h。反应结束后,加入水(10ml),浓盐酸调节ph至3,乙酸乙酯萃取,无水硫酸钠干燥,旋蒸除去溶剂,得粗品化合物2。将粗品化合物2直接溶于乙醇(100ml),升温至80℃,逐滴滴加浓硫酸(4ml),继续反应5h。反应结束后,加入10%氢氧化钠水溶液调节ph至12,乙酸乙酯萃取,无水硫酸钠干燥,旋蒸除去溶剂,以石油醚和二氯甲烷作为洗脱剂进行柱层析提纯得纯品化合物3(4.3g,36.6%)。将化合物3(4.3g,26.8mmol)溶于乙酸酐(30ml),加入丙二腈(2.7g,40.87mmol),140℃搅拌过夜。反应结束后,旋蒸除去溶剂,以二氯甲烷作为洗脱剂进行柱层析得亮黄色固体ewgo(2.3g,41.2%)。
2)使用与ewgo相同的合成方法合成化合物7,化合物7(3.0g,8.1mmol)以乙酸乙酯(50ml)作为溶剂,加入钯炭(0.3g),使用氩气将反应瓶内的空气置换3次,插上氢气囊,反应12h。反应结束后,过滤,旋蒸除去溶剂,以石油醚和乙酸乙酯进行柱层析得到黄色固体化合物8(2.17g,95.6%)。化合物8(2.17g,7.74mmol)溶于乙腈(20ml),加入碳酸钾(2.14g,15.48mmol),升温至80℃,逐滴滴加炔丙溴(1.38g,11.6mmol),滴加完毕后,继续反应8h。反应结束后,过滤,旋蒸除去溶剂,以石油醚和二氯甲烷进行柱层析得黄色固体alkyneewgo(2.2g,89.6%)。1hnmr(400mhz,cdcl3)δ(ppm)8.83(s,1h),6.95(s,1h),6.65(d,j=0.9hz,1h),4.85(d,j=2.4hz,2h),2.59(t,j=2.4hz,1h),2.40(d,j=0.8hz,3h),1.43(s,9h).13cnmr(100mhz,cdcl3)δ(ppm)161.37,160.92,153.37,153.20,138.68,124.10,117.26,116.16,110.93,105.28,100.91,76.92,76.81,59.75,56.09,36.00,29.67,20.34.
使用与alkyneewgo相同合成方法得到azideewgo。1hnmr(400mhz,cdcl3)δ(ppm)8.82(s,1h),6.82(s,1h),6.63(d,j=0.4hz,1h),4.21(t,j=5.2hz,2h),3.81(t,j=5.2hz,2h),2.40(d,j=0.4hz,3h),1.45(s,9h).13cnmr(100mhz,cdcl3)δ(ppm)162.21,160.82,153.32,138.36,124.12,117.23,116.11,110.76,105.27,100.18,99.98,66.79,59.78,50.25,36.02,29.64,20.29.
实施例二:合成ewgs、alkyneewgs,合成路线见附图3
1)将磷酸(22.5g,229.6mmol)和五氧化二磷(40.7g,287.0mmol)在90℃搅拌一小时,对叔丁基苯硫酚(4.5ml,26.1mmol)和乙酰乙酸乙酯(3.3ml,26.1mmol)的混合物被逐滴滴加,继续反应2h。反应结束后,将反应液直接倒入冰水中,10%氢氧化钠水溶液调节ph至9,乙酸乙酯萃取,无水硫酸钠干燥,旋蒸除去溶剂,以石油醚和二氯甲烷为洗脱剂进行柱层析得到纯品黄色油状化合物10(3.09g,51.0%)。将化合物10(2g,8.6mmol)溶解在15ml乙酸酐中,加入丙二腈(0.68g,10.3mmol),140℃反应过夜,反应结束后,旋蒸除去溶剂,以石油醚和二氯甲烷为洗脱剂进行柱层析,得黄色固体ewgs(1.12g,46.4%)。1hnmr(400mhz,cdcl3)δ8.96(d,j=2.0hz,1h),7.69(dd,j=8.8,2.0hz,1h),7.58(d,j=8.4hz,1h),7.41(d,j=0.8hz,1h),2.52(d,j=0.4hz,3h),1.41(s,9h).13cnmr(100mhz,cdcl3)δ156.53,152.27,148.81,132.94,129.98,126.68,125.21,124.62,120.58,117.38,116.08,67.36,35.70,31.09,23.51.
2)使用与化合物10相同的合成方法合成化合物12,化合物12(4.5g,21.8mmol)溶于二氯甲烷中,三溴化硼(6ml,63.5mmol)在0℃下被逐滴滴加,逐渐升至室温,搅拌过夜。反应结束后,加入冰水淬灭多余的三溴化硼,除去二氯甲烷,用乙酸乙酯萃取,无水硫酸钠干燥,旋蒸除去溶剂,以石油醚和二氯甲烷作为洗脱剂进行柱层析得白色固体化合物13(3.4g,81.0%)。化合物13(4.4g,22.9mmol),碳酸钾(7.9g,57.2mmol)加入丙酮(50ml)中,升温至80℃,逐滴滴加炔丙溴(2.7ml),反应8h。过滤,旋蒸除去丙酮,以石油醚和二氯甲烷为洗脱剂进行柱层析得白色固体化合物14(4.59g,87%)。化合物14(3.4g,14.78mmol)和丙二腈(1.3g,19.7mmol)溶于乙酸酐(20ml),140℃搅拌过夜,旋蒸除去溶剂,以石油醚和二氯甲烷为洗脱剂进行柱层析得得alkyneewgs(1.75g,42.5%)。1hnmr(400mhz,dmso-d6)δ(ppm)8.38(d,j=2.4hz,1h),8.00(d,j=8.8hz,1h),7.56(dd,j=8.8,2.4hz,1h),7.38(s,1h),4.93(d,j=2.0hz,2h),3.69(t,j=2.hz,1h),2.60(s,3h).13cnmr(100mhz,dmso-d6)δ(ppm)157.21,155.58,151.89,129.65,129.00,125.64,121.86,119.83,117.91,116.53,112.42,79.72,78.55,65.19,56.69,23.49.
实施例三:合成ewgse、alkyneewgse、azideewgse,合成路线见附图3
1)将邻溴苯甲酰氯(25g,0.114mol)溶于干燥的四氢呋喃(100ml)和三乙胺(100ml)的混合溶液,除去反应液中的空气,加入二氯二(三苯基磷)钯(0.8g,11.4mmol)和碘化亚铜(259.7mg,1.36mmol),氩气保护,冰水浴下逐滴滴加丙炔(0.114mol,1m的四氢呋喃溶液)。滴加完毕后,撤掉冰水浴,慢慢恢复至室温,继续反应17h。加入甲醇(10ml)淬灭反应,旋蒸除去溶剂,乙酸乙酯溶解粗品,5%硫酸水溶液、碳酸氢钠水溶液、饱和氯化钠水溶液分别洗涤2次,无水硫酸钠干燥,旋蒸除去溶剂,以石油醚和乙酸乙酯为洗脱剂进行柱层析,纯化得淡黄色油状化合物17(7.27g,28.6%)。硒粉(1.54g,19.5mmol)和硼氢化钠(0.54g,14.27mmol)溶于dmf(60ml),100℃反应1h,氩气保护下缓慢滴加化合物18(2.03g,9.1mmol)的乙醇溶液,滴加完毕后,继续反应5h,反应结束后,旋蒸除去dmf,溶于乙酸乙酯,饱和食盐水洗涤3次,干燥,旋蒸除去溶剂,以石油醚和乙酸乙酯进行柱层析纯化得白色固体化合物18(1.32g,65.0%)。1hnmr(400mhz,cdcl3)δ(ppm)8.56(d,j=7.2hz,1h),7.63-7.54(d,j=6.8hz,1h),7.53-7.41(m,2h),6.97(s,1h),2.51(s,3h).13cnmr(100mhz,cdcl3)δ(ppm)182.48,152.40,136.56,131.57,131.41,129.99,128.15,127.56,127.03,25.04.
将化合物18(2.23g,10.4mmol)和丙二腈(0.79g,12mmol)溶于乙酸酐(15ml)中,140℃反应过夜。旋蒸除去溶剂,以石油醚和二氯甲烷为洗脱剂进行柱层析得黄色固体ewgse(1.21g,42.9%)。1hnmr(400mhz,cdcl3)δ(ppm)8.73(d,j=4.8hz,1h),7.71(d,j=4.4hz,1h),7.52(m,3h),2.60(s,3h).13cnmr(100mhz,cdcl3)δ(ppm)158.83,151.34,135.45,131.61,129.77,129.15,128.19,125.97,122.37,116.63,115.31,72.36,25.32.
2)使用与化合物18相同的方法合成化合物21,化合物21(2.5g,9.88mmol)以二氯甲烷(20ml)为溶剂,0℃下逐滴滴加三溴化硼(2.75ml,29.09mmol),逐渐升至室温,搅拌过夜,反应结束后,加入冰水淬灭多余的三溴化硼,用乙酸乙酯萃取,无水硫酸钠干燥,旋蒸除去溶剂,以乙酸乙酯和二氯甲烷为洗脱剂进行柱层析得白色固体化合物22(1.96g,83.0%)。化合物22(3.7g,15.47mmol),溶于乙腈(50ml),加入碳酸钾(4.27g,30.90mmol)升温至80℃,逐滴滴加炔丙溴(2.7g,22.7mmol),滴加完毕,继续反应8h。反应结束后,过滤,旋蒸除去溶剂,以石油醚和二氯甲烷进行柱层析得白色固体化合物23(3.3g,77.0%)。使用与化合物23相同的合成方法得到白色固体化合物24。将化合物23(2g,7.2mmol)溶于20(ml)乙酸酐中,加入丙二腈(0.57g,8.6mmol),140℃搅拌过夜,反应结束后,旋蒸除去溶剂,以石油醚和二氯甲烷为洗脱剂进行柱层析,得黄色固体化合物alkynepsse(0.99g,42.0%)。1hnmr(400mhz,dmso-d6)δ(ppm)8.26(d,j=2.8hz,1h),8.07(d,j=8.8hz,1h),7.53-7.40(m,2h),4.91(d,j=2.0hz,2h),3.67(t,j=2.0hz,1h),2.65(t,j=6.4hz,3h).13cnmr(101mhz,dmso-d6)δ(ppm)158.29,157.09,155.18,131.50,128.42,126.54,121.74,121.10,117.54,116.14,114.42,79.62,78.70,69.90,56.70,25.39.
使用与alkynepsse相同的合成方法合成黄色固体化合物azidepsse。1hnmr(400mhz,cdcl3)δ(ppm)8.21(d,j=2.8hz,1h),8.04(d,j=8.8hz,1h),7.47(d,j=0.8hz,1h),7.39(dd,j=8.8,2.8hz,1h),4.28(t,j=4.8hz,2h),3.75(t,j=5.2hz,2h),2.65(s,3h).13cnmr(100mhz,cdcl3)δ(ppm)158.45,157.77,155.08,131.51,128.23,126.57,121.71,121.57,117.69,116.16,113.32,69.89,67.82,49.80,25.35.
实例四:合成pso-br、pso-i、alkynepso-i,合成路线见附图4
1)3,5-二溴-4-苯甲醛(0.8g,2.9mmol)溶于乙腈(20ml),分别加入哌啶(1.6ml)和乙酸(0.8ml),再加入适量的分子筛,搅拌,升温至85℃,加入ewgo(0.5g,2.4mmol),继续反应8h。反应结束后,过滤除掉分子筛,加入乙酸乙酯(50ml),1m的盐酸水溶液洗涤,过滤得粗品,以二氯甲烷为洗脱剂通过柱层析的方式进行纯化,得黄色固体化合物pso-br(0.42g,37.4%)。1hnmr(400mhz,dmso-d6)δ(ppm)10.57(s,1h),8.73(d,j=8.4hz,1h),8.02(s,2h),7.94(m,1h),7.72(d,j=8.4hz,1h),7.63(m,2h),7.49(d,j=15.6hz,1h),6.98(s,1h).
2)使用和pso-br相同的合成方法合成pso-i。1hnmr(400mhz,dmso-d6)δ(ppm)10.10(s,1h),8.72(d,j=8.4hz,1h),8.18(s,2h),7.93(m,1h),7.72(d,j=8.4hz,1h),7.64-7.56(m,2h),7.44(d,j=16.0hz,1h),6.98(s,1h).13cnmr(101mhz,dmso-d6)δ(ppm)158.60,157.95,153.30,152.44,139.40,136.18,135.90,131.37,126.60,125.12,119.39,119.03,117.68,117.57,116.32,107.04,87.85,60.44.hrms(esi):m/z[m+h]+calcdforc20h11i2n2o2564.8905;found564.8819.
3)使用和pso-br相同的合成方法合成alkynepso-i。1hnmr(400mhz,dmso-d6)δ(ppm)10.05(s,1h),8.66(s,1h),8.16(s,2h),7.52(d,j=16.0hz,1h),7.42(d,j=16.0hz,1h),7.33(s,1h),6.90(s,1h),5.10(d,j=2.hz,2h),3.73(s,1h),1.40(s,9h).13cnmr(101mhz,dmso-d6)δ(ppm)161.75,157.94,157.44,153.14,152.85,139.21,137.75,135.10,131.67,123.25,119.41,118.09,116.64,110.69,106.86,102.22,87.59,79.70,78.44,58.28,56.92,55.37,36.00.
实例五:合成pss-br、pss-i,合成路线见附图4
1)使用和pso-br相同的合成方法合成pss-br。1hnmr(400mhz,dmso-d6)δ(ppm)10.49(s,1h),8.80(s,1h),8.05(s,2h),7.91(m,2h),7.76(d,j=16.0hz,1h),7.63(s,1h),7.34-7.25(d,j=16.8hz,1h),1.36(d,j=2.2hz,9h).13cnmr(101mhz,dmso-d6)δ(ppm)156.23,152.45,151.80,148.31,134.37,132.23,131.95,131.09,130.24,128.14,126.27,124.63,124.56,121.78,118.11,116.43,112.66,66.76,35.80,31.11.
2)使用和pso-br相同的合成方法合成pss-i。1hnmr(400mhz,dmso-d6)δ(ppm)10.01(s,1h),8.80(d,j=1.2hz,1h),8.22(s,2h),7.94-7.86(m,2h),7.71(d,j=16.4hz,1h),7.64(s,1h),7.27(d,j=16.0hz,1h),1.36(s,9h).13cnmr(101mhz,dmso-d6)δ(ppm)157.20,156.13,155.96,151.67,148.32,139.07,134.00,131.90,131.69,130.94,130.85,128.03,125.74,124.53,121.56,118.09,116.41,87.51,66.51,35.75,31.13.hrms(esi):m/z[m+h]+calcdforc24h19i2n2os636.9302;found636.9291.
实例五:合成psse、psse-br、psse-i、azidepsse-i,合成路线见附图4
1)使用和pso-br相同的合成方法合成psse。1hnmr(400mhz,dmso-d6)δ(ppm)10.06(s,1h),8.61(d,j=7.6hz,1h),8.09(d,j=6.8hz,1h),7.80-7.50(m,6h),7.31-7.16(d,j=16.0hz,1h),6.83(d,j=6.8hz,2h).13cnmr(100mhz,dmso-d6)δ(ppm)160.07,159.07,151.84,139.03,134.12,132.58,130.56,130.47,129.58,128.59,126.85,126.51,125.04,122.45,117.64,116.38,116.17,70.49.hrms(esi):m/z[m+h]+calcdforc20h13n2ose377.0188;found377.0181.
2)使用和pso-br相同的合成方法合成psse-br。1hnmr(400mhz,dmso-d6,figures23)δ(ppm)10.48(s,1h),8.60(d,j=8.0hz,1h),8.13-8.01(m,3h),7.83-7.61(m,4h),7.19(d,j=16.0hz,1h).13cnmr(100mhz,dmso-d6,figures24)δ(ppm)159.04,152.37,150.70,135.53,134.10,132.69,132.26,130.60,130.52,129.68,128.70,128.31,126.39,123.83,117.43,115.92,112.72,71.80.hrms(esi):m/z[m]calcdforc20h10br2n2ose533.0810;found533.0991.
3)使用和pso-br相同的合成方法合成psse-i。1hnmr(400mhz,dmso-d6)δ(ppm)10.01(s,1h),8.59(dd,j=8.0,1.2hz,1h),8.22(s,2h),8.10(dd,j=7.6,1.2hz,1h),7.81-7.59(m,4h),7.16(d,j=16.4hz,1h).13cnmr(100mhz,dmso-d6)δ(ppm)159.10,157.16,150.95,139.13,135.37,134.12,132.66,132.08,130.59,129.68,128.68,127.88,126.42,123.65,117.47,115.95,87.65,71.65.hrms(esi):m/z[m+h]+calcdforc20h11i2n2ose628.8121;found628.8042.
4)使用和pso-br相同的合成方法合成azidepsse-i。1hnmr(400mhz,dmso-d6)δ(ppm)9.69(s,1h),8.17(m,3h),8.01(d,j=8.8hz,1h),7.68(m,2h),7.36(dd,j=8.4,1.6hz,1h),7.11(d,j=15.6hz,1h),4.27(t,j=4.4hz,2h),3.75(t,j=3.6hz,2h).13cnmr(100mhz,dmso-d6)δ(ppm)158.44,157.78,151.71,139.17,135.36,131.79,131.31,127.39,127.23,125.89,122.99,121.56,117.92,116.27,113.43,88.01,70.25,67.80,49.78.
以上所述仅是本发明的优选实施方式,应当指出,对于使用本技术领域的研究人员,在不脱离本发明技术原理的前提下,对这些实施例进行的多种修改,这些修改应该视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!