本发明涉及生物工程领域,尤其涉及一种增强子筛选表达载体及利用随机序列筛选增强子的方法。
背景技术:
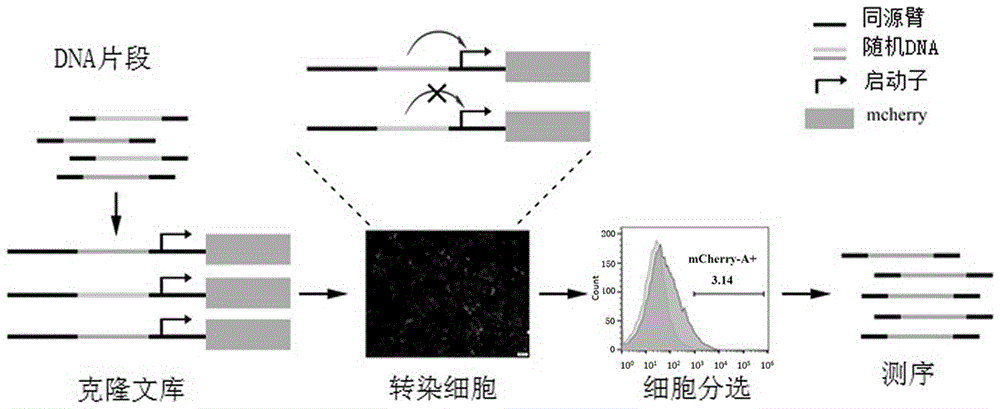
:真核生物的基因表达是一个精细,复杂且处于动态变化的过程,它受到基因组转录因子,表观遗传修饰各种调控元件等多因素的复杂调控从而执行各种基因时空特异性表达。其中顺式调控元件通过招募不同的反式作用因子使染色质高级结构发生变化,从而启动或沉默基因的表达,是近些年基因调控研究的热门领域,因此,对于基因组中顺式调控元件的鉴定对于深入理解基因表达调控的机制具有重要意义。增强子是一类具有重要作用的顺式调控元件,对基因的表达调控具有重要作用。对增强子的基因组特征进行描述是dna元件百科全书项目(encode)的重要内容。为此,现在已经开发出了很多方法,如染色质免疫沉淀(chip-seq),dnasei高敏感位点测序(dnase-seq),甲醛辅助分离调控元件技术,染色质开放性测序技术(atac-seq)以及starr-seq(自我转录活性调控区测序),总的来说,这些技术主要是依靠组蛋白修饰或染色质可获得性的染色质特征,并认为这些是增强子位点的基本特征,但是由于染色质特征的复杂性以及分析方法的原因,这种对基因组中增强子的鉴定不是很准确。目前,已经定义的增强子元件大多数在几百个碱基对(bp)以上,例如h3k27acchip-seq的覆盖度范围为200-60kbp(如新定义的超级增强子),如此庞大的区域可能包含多个信号。同时,通过对这些不同基因组数据集的motif分析,我们发现每个位点的核心区域仅为几个或几十bp长度。众所周知,由于染色体中核小体本身结构的特征这些传统的方法无法做到比147bp更短的序列,用现有的技术去鉴定基因组中的核心增强子是一件几乎不可能实现的事情。因此,现有技术有待于进一步发展。技术实现要素:本发明目的是针对上述问题,提供一种增强子筛选表达载体及利用随机序列筛选增强子的方法。为了实现上述目的,本发明的技术方案是:一种增强子筛选表达载体,其中,该增强子筛选表达载体用于对随机序列中活性增强子进行筛选,所述增强子筛选表达载体由穿梭质粒载体改造而成,所述增强子筛选表达载上的核心部件包括mini启动子及其下游的mcherry报告基因,mini启动子上游包含随机序列的重组位点。一种利用随机序列筛选核心增强子的方法,其中,所述方法包括以下步骤:s1、合成随机序列文库;s2、利用xhoi酶和hindiii酶对增强子筛选表达载体进行酶切,得到线性载体。s3、利用线性载体和随机序列文库构建质粒文库;s4、将所构建的质粒文库转染细胞,并基于荧光表达情况,通过流式富集增强子阳性细胞;s5、对所富集的增强子阳性细胞进行dna提取,并利用所提取的dna建库再进行高通量测序,所述利用随机序列筛选核心增强子的方法,其中,随机序列文库中序列长20-50bp。所述利用随机序列筛选核心增强子的方法,其中,随机序列文库使用前进行稀释,具体稀释条件为:12000g,rt,离心90s,加无菌水稀释成浓度为50ng/ul的溶液。所述利用随机序列筛选核心增强子的方法,其中,步骤s2中酶切体系为:nebuffer2.1,5ul;xhoi,2ul;hindiii,2ul;dna,20ug,加双蒸水至体系为50ul。所述利用随机序列筛选核心增强子的方法,其中,所述步骤s3具体包括:利用同源重组酶对线性载体和随机序列文库进行同源重组,并将重组质粒加入到感受态细胞中,之后利用质粒提取试剂盒提取重组质粒。所述利用随机序列筛选核心增强子的方法,其中,所述步骤s4具体包括:从液氮中取出细胞293t,空气中放置10-15s,在37℃水浴锅中快速解冻,将细胞悬液转移到离心管中,200g,rt,离心5min;弃上清,加完全培养基重悬细胞,然后置于培养箱中;待细胞生长到80%-90%汇合度时,使用脂质体试剂转染细胞,48h后观察荧光表达情况,其中,每个100mm培养皿加入2ml消化酶消化细胞2min,加入2ml完全培养基终止消化,200g,rt,离心5min;弃上清,加入5mldpbs洗涤一次,再次离心收集细胞;利用流式分选细胞仪富集增强子阳性细胞。所述利用随机序列筛选核心增强子的方法,其中,所述步骤s5中对所富集的增强子阳性细胞进行dna提取具体为:将富集的增强子阳性细胞2000g,rt,离心5min,弃上清;在细胞沉淀中加入600ul细胞裂解液,70ul10%sds,5ul蛋白酶k,轻轻上下颠倒数下,混匀后放在56℃的烘箱内孵育1h,其间不间断轻轻振荡使其消化完全;加入等体积平衡酚,上下轻轻颠倒混匀,13000g,离心10min,吸取上清液于新的ep管里;加入等体积氯仿:异戊醇混合液,上下轻轻颠倒混匀,13000g,rt,离心10min,吸取上清液于新的ep管内;在上清液中加入两倍体积的-20℃冰冻无水乙醇,于-20℃沉淀至少1h,13000g,rt,离心15min,弃上清液;加入500μl的75%冰乙醇,混匀漂洗以除去残余的盐,13000g,rt,离心2min,吸去上清液;将管倒立在卷纸上,晾干dna沉淀块,除去残余乙醇;加入适量te缓冲液或灭菌双蒸水溶解dna沉淀,保存以备电泳检测。所述利用随机序列筛选核心增强子的方法,其中,所述步骤s5中利用所提取的dna建库具体包括:根据载体设计扩增引物,并利用高效扩增酶以及pcr扩增程序扩增基因组dna,再进行切胶回收纯化目的条带;将第一步的胶回收产物利用建库试剂盒进行扩增,并在扩增的产物加上测序用接头。所述利用随机序列筛选核心增强子的方法,其中,所述利用设计的扩增引物扩增基因组dna的pcr扩增程序为:98℃45s,98℃10s,60℃10s,72℃5s,72℃1min,共7个循环。所述利用随机序列筛选核心增强子的方法,其中,利用建库试剂盒扩增的产物加入测序用接头进行连接,在接接后产物中加入1.5倍体积dnacleanbeads,使用移液器吹打10次混匀,室温孵育5min;将反应管短暂离心并置于磁力架中静置,待溶液澄清,移除上清;保持ep管处于磁力架中,加入200μl新鲜配制的80%乙醇漂洗磁珠,室温孵育30s,移除上清,重复此步骤一次;保持ep管处于磁力架中,开盖空气干燥磁珠3-5min;将ep管从磁力架中取出,加入相应体积灭菌超纯水洗脱,涡旋振荡或使用移液器轻轻吹打充分混匀,于室温放置2min,将反应管短暂离心并置于磁力架中静置,待溶液澄清,吸取上清至新ep管中备用;将dnacleanbeads纯化后的产物用kapalibraryamplicationprimermix扩增,琼脂糖电泳,再进行切胶回收,对回收产物检测荧光浓度后进行高通量测序。有益效果:本发明提供一种增强子筛选表达载体及利用随机序列筛选增强子的方法,该方法利用增强子筛选表达载体和筛选文库实现对增强子序列的有效鉴别,相较于现有技术,该方法对基因组中增强子的鉴定准确率大幅提升,同时该方法能够针对几十bp序列中的增强子进行有效鉴定很大程度上提高了精确性,对于深入理解基因表达调控的机制具有重要意义。附图说明图1为本发明实验原理流程图;图2为本发明本发明具体实施例中增强子筛选表达载体图谱;图3为本发明具体实施例中利用xhoi和hindiii对增强子筛选表达载体酶切后琼脂糖凝胶电泳检图;图4为本发明具体实施例中同源重组质粒pmx-mp-n25-mchery琼脂糖凝胶电泳检图;图5为本发明随机序列文库转染细胞荧光图。图6为本发明细胞流式筛选富集结果。图7为本发明两步法建库测序琼脂糖凝胶电泳检图。具体实施方式下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。如图1所示,本发明将随机合成序列(20-50bp)同源重组到mini启动子的上游,如果随机序列有增强子活性将激活下游报告基因mchery的表达,通过流式富集把增强子阳性细胞筛选下来,通过建库再去高通量测序得到增强子序列。本发明所用增强子筛选表达载体用于对随机序列中活性增强子进行筛选,所述增强子筛选表达载体由穿梭质粒载体改造而成,所述增强子筛选表达载上的核心部件包括mini启动子及其下游的mcherry报告基因,mini启动子上游包含随机序列的重组位点,本发明增强子筛选表达载体(pmx-mchery)具体图谱如图2所示。具体实验过程如下:1、随机序列文库的合成:25bp长随机序列文库委托金唯智生物科技有限公司合成。随机文库稀释:12000g,rt,离心90s,加无菌水稀释成浓度为50ng/ul的溶液。2、载体线性化2.1载体双酶切利用xhoi(neb.cat.no.r0146v)和hindiii(neb.cat.no.r0104b)在nebuffer2.1(neb.cat.no.b7202s)缓冲液中对pmx-mchery载体(图2)进行酶切,酶切后序列电泳图如图3所示,酶切体系如下:组分用量nebuffer2.15ulxhoi2ulhindiii2uldna20ugddwaterto50ul2.2酶切载体胶回收称取0.5g胶粉溶于50ul1xtae中,微波炉加热1分钟,拿出摇晃数下继续加热1min,倒入胶板中静置30min。以超螺旋dnaladder(neb;cat.no.n0472s)为对照,每孔200-500ng上样量,设置参数120v,130ma,45min进行琼脂糖电泳;用胶回收试剂盒(zymo;cat.no.d4008)回收目的条带。质粒文库构建3.1同源重组质粒(pmx-mp-n25-mchery)构建利用同源重组酶(vazyme;cat.no.c112)对线性载体和随机文库进行同源重组,每个反应随机文库10ng,线性载体120ng,37℃反应30min,将重组产物加入到感受态细胞(tsingke;cat.no.tsc01)中,轻轻混匀,冰上静置30min,42℃水浴45s,立即取出冰上静置2-3min,每管感受态加入500ulsoc培养基在37℃,200rpm摇床中活化1h,取出后在离心机中2000g,rt,5min,弃上清,留100ulsoc重悬菌体,将菌液均匀涂布在固体培养板上,37℃培养箱过夜培养(具体时间视感受态效率而定)。3.2质粒提取在固体培养皿中添加lb培养基将菌落全部刮下,并补充适当体积的lb培养基(添加amp)到锥形瓶中,设置摇床参数37℃,200rpm摇菌12-16小时。第二天用质粒提取试剂盒(omega;cat.no.d6950)提取质粒,琼脂糖凝胶电泳检测质粒质量(图4)。细胞培养从液氮中取出细胞(293t),空气中放置10-15s,在37℃水浴锅中快速解冻,将细胞悬液转移到离心管中,200g,rt,离心5min;弃上清,加完全培养基(90%dmem(thermfisher;cat.no.10566024)+10%fbs(gibco;cat.no.10437028))重悬细胞,然后置于培养箱中。细胞转染及增强子阳性细胞富集待细胞生长到80%-90%汇合度时,使用脂质体试剂(promega;e2311)转染细胞,同时分别设置pmx-mp-mchery和pmx-sp40-mp-mchery为阴性和阳性对照。48h后观察荧光表达情况(图5),每个100mm培养皿加入2ml消化酶(thermofisher;cat.no.a1110501)消化细胞2min,加入2ml完全培养基终止消化,200g,rt,离心5min;弃上清,加入5mldpbs洗涤一次,再次离心收集细胞;利用流式分选细胞仪富集增强子阳性细胞(图6)。测序建库6.1阳性细胞基因组dna提取将富集的细胞2000g,rt,离心5min,弃上清;在细胞沉淀中加入600ul细胞裂解液(ste),70ul10%sds(终浓度为1%),5ul蛋白酶k(终浓度为100~200ug/ml),并轻轻上下颠倒数下,混匀后放在56℃的烘箱内孵育1h,其间要不间断轻轻振荡使其消化完全。抽提步骤1:加入等体积平衡酚(约700μl),上下轻轻颠倒混匀,13000g,离心10min,小心吸取上清液于新的ep管里。抽提步骤2:加入等体积氯仿:异戊醇(24:1)混合液,上下轻轻颠倒混匀,13000g,rt,离心10min,小心吸取上清液于新的ep管内。如中间还有白色蛋白层,重复抽提步骤1、2,至无白色蛋白层。沉淀:在上清液中加入两倍体积的-20℃冰冻无水乙醇,于-20℃沉淀至少1h,13000g,rt,离心15min,弃上清液。注意此步需要小心操作以防止dna沉淀块被一起丢弃。漂洗:加入500μl的75%冰乙醇,混匀漂洗以除去残余的盐,13000g,rt,离心2min,吸去上清液。若杂质较多,可以重复该步骤几次。干燥:将管倒立在卷纸上,晾干dna沉淀块,除去残余乙醇。溶解:加入适量te(ph8.0)或灭菌双蒸水溶解dna沉淀,保存以备电泳检测。6.2建库本发明建库分为两步pcr建库。第一步:根据载体设计扩增引物(本实验:pmx-ty1:5’—tgcaggtgccagaacatttc—3’pmx-ty2:5’—gtggctttaccaacagtacc—3’)利用高效扩增酶(kapa;cat.no.kk2601)以及pcr扩增程序(98℃45s,98℃10s,60℃10s,72℃5s,72℃1min)扩增基因组dna(7循环),再进行切胶回收纯化目的条带(图7)。第二步:将第一步的胶回收产物利用建库试剂盒(kapa;cat.no.kk8504)进行endrepairanda-tailing,体系如下:按以下程序进行扩增:对上述扩增的产物加上测序用的接头(vazyme;cat.no.n805),体系如下:将上述体系在pcr仪中20℃反应15分钟。在上述混合体系加入1.5倍体积dnacleanbeads(beckman;cat.no.a63881)使用移液器吹打10次混匀,室温孵育5min;将反应管短暂离心并置于磁力架中静置,待溶液澄清(约5min),移除上清。保持ep管处于磁力架中,加入200μl新鲜配制的80%乙醇漂洗磁珠。室温孵育30s,移除上清,重复此步骤一次。保持ep管处于磁力架中,开盖空气干燥磁珠3-5min(不能使磁珠皲裂)。将ep管从磁力架中取出,加入相应体积灭菌超纯水洗脱。涡旋振荡或使用移液器轻轻吹打充分混匀,于室温放置2min。将反应管短暂离心并置于磁力架中静置,待溶液澄清(约5min),小心吸取上清至新ep管中备用。将beads纯化后的产物用kapalibraryamplicationprimermix按照第一步的扩增程序扩增4个循环,120v,130ma琼脂糖电泳35min,再进行切胶回收(图7),对回收产物qubit检测浓度后进行高通量测序。可以理解的是,对本领域普通技术人员来说,可以根据本发明的技术方案及本发明构思加以等同替换或改变,而所有这些改变或替换都应属于本发明所附的权利要求的保护范围。当前第1页1 2 3