通过内含子促进人类胚胎细胞因子-1在293T中表达的方法与流程
本发明涉及一种增强子序列、一种质粒载体pnc1、该质粒载体pnc1的制备方法、该质粒载体pnc1在表达fgf-2中的用途,以及一种转化体。
背景技术:
fgf2是制药和保健行业中非常有价值的蛋白质。fgf2是治疗神经退行性疾病,心脏病,血管生成,难以愈合的伤口和骨折的有效治疗性蛋白质;它还在干细胞的大规模生产中发挥重要作用。
由于制备成本低,复制率高,生产率高,大肠杆菌长期用于纯化重组蛋白[1-4]。然而,由于外源蛋白质引起的毒性,缺乏正确的蛋白质折叠和翻译后修饰,使用原核表达系统在纯化哺乳动物蛋白质时经常遇到障碍[5,6]。多年来,科学家一直在使用真核宿主如酵母和昆虫细胞来克服这些局限[7]。尽管如此,使用人类来源的培养细胞进行人类来源的蛋白质表达似乎是直观的。实际上,出现了使用哺乳动物细胞产生重组蛋白的上升趋势[8,9]。
目前,fgf2制备和纯化最广泛使用的宿主系统之一是细菌。然而,简单的原核生物缺乏必要的翻译后修饰,包括剪接,糖基化和二硫键,用于纯化的蛋白质的活性和溶解度。纯化蛋白质的天然折叠对其功能和溶解性至关重要。当在细菌系统中表达真核蛋白时经常发现蛋白质聚集或包涵体。包括在较低温度下诱导蛋白质或使蛋白质聚集体变性和复性的方法并不总是产生良好的产率。因此,制备和纯化具有生物活性的fgf2极其昂贵(4000美元/毫克),这阻碍了其广泛的应用。
技术实现要素:
为了克服现有技术的缺陷,本发明一方面提供了一种增强子,该增强子能够在哺乳动物细胞中大大增加具有生物活性的fgf2的表达。
另一方面,本发明提供了含有所述增强子的质粒载体pnc1。
另一方面,本发明提供了本发明质粒载体pnc1的制备方法。
另一方面,本发明提供了本发明质粒载体pnc1在表达fgf-2中的应用。
另一方面,本发明提供了含有本发明质粒载体pnc1的转化体。
因此,本发明提供了一种增强子序列,其特征在于,该增强子序列包括nf-κb结合位点的dna序列和creb结合位点的dna序列。
本发明还提供了一种质粒载体pnc1,其特征在于,该质粒载体包括本发明所提供的增强子序列。
本发明还提供了一种质粒载体pnc1的制备方法,其特征在于,该方法包括以下步骤:
a.合成含有nf-κb结合位点的dna序列和creb结合位点的dna序列的dna序列,其中,nf-κb结合位点的dna序列的末端带有nhei限制性酶切位点,creb结合位点的dna序列的末端带有hindiii限制性酶切位点;
b.用nhei和hindiii消化原始质粒载体,回收大片段;
c.采用连接酶连接步骤a获得的dna序列与步骤b获得的大片段;
d.将步骤c获得的连接产物转染细胞,筛选阳性克隆,提取质粒。
本发明提供的质粒载体pnc1能够显著提高fgf-2的表达水平,而且表达的fgf-2基本上是可溶的,其生物活性与商购的fgf-2的生物活性相当。因此,采用本发明提供的质粒载体pnc1表达fgf-2,能显著提高具有生物活性fgf-2的表达,从而能大大降低制备和纯化fgf-2的成本。此外,质粒载体pnc1可用于表达多种有价值的蛋白质。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本申请的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
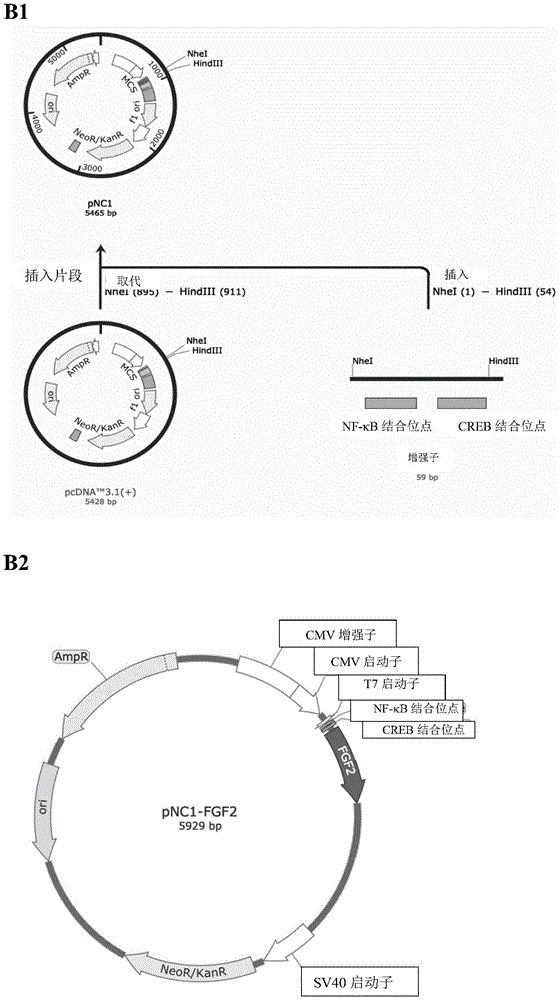
图1是fgf2构建体和his-dnae-fgf2构建体的示意图。(a)合成fgf2基因的成熟功能片段的dna,并克隆到在cmv增强子/启动子的控制下的pcdna3.1(+)载体中,形成pcdna3.1-fgf2构建体。(b1)合成nf-κb结合位点和creb结合位点并克隆在cmv启动子后,形成pnc1载体;(b2)然后将fgf2基因的成熟功能片段的dna克隆到pnc1载体中,形成pnc1-fgf2构建体。(c)通过融合pcr将6xhis标签和npudnae内含肽基因与fgf2融合,并克隆到pnc1载体中,形成pnc1-6xhis-dnae-fgf2构建体。
图2是成熟功能片段fgf2蛋白在hek293t细胞中的表达。(a)将pcdna3.1-fgf2和pnc1-fgf2构建体转染到hek293t细胞中,并在指定的时间点通过蛋白质印迹分析全细胞裂解物。(b)为(a)相对β-肌动蛋白标准化的光密度分析。pnc1-fgf2构建体在24小时,36小时和48小时显示出比pcdna3.1-fgf2构建体更高的表达水平(**p<0.01,***p<0.001;在每个时间点n=5)。(c)通过蛋白质印迹分析来自细胞裂解物的上清液(s)和沉淀(p)。商购fgf2(+ve)作为阳性对照。对于pcdna3.1-fgf2和pnc1-fgf2构建体,hek293t表达的fgf2都是相当可溶的。
图3是在hek293t细胞中表达的fgf2的纯化。(a)将全细胞裂解物和来自尺寸排阻色谱样品的洗脱物进行sds-page并用考马斯亮蓝染色。(b)将来自(a)的样品冻干并在0.1xpbs中重构,然后在sds-page后通过银染色分析。在纯化的fgf2中仅观察到1个弱带(由箭头指示)。
图4是纯化的fgf2的生物测定。(a)mtt测定以测量fgf2相对于pbs对照对于c2c12细胞的细胞活力的影响。在指定的时间点可以观察到在培养基中1ng/ml的商购和纯化的fgf2都能够诱导细胞生长(n=3)。(b)将浓度为2ng/ml的商购和纯化的fgf2加入培养基中3天。在第0天和第3天拍摄相差显微照片。在fgf2处理3天后观察到pc12细胞中的延长的神经突生长(用箭头表示)。
图5是成熟功能片段fgf2蛋白在hek293t细胞中的表达以及通过内含肽辅助切割纯化fgf2。将pnc1-6xhis-dnae-fgf2构建体转染到hek293t细胞中,收集培养基并以2,000g旋转10分钟以除去细胞碎片,并通过0.45μm过滤器过滤。通过ni-nta亲和层析纯化蛋白质。在切割缓冲液中诱导dnae内含肽的切割。从诱导后2小时开始,将全长his-dnae-fgf2蛋白(由*表示的条带)切割成fgf2(用#表示的条带)。诱导后6小时后裂解完成。
图6是纯化的和内切蛋白切割的fgf2的生物测定。(a)mtt测定以测量fgf2相对于pbs对照对于c2c12细胞的细胞活力的影响。在指定的时间点观察到在培养基中1ng/ml的纯化和内切蛋白切割的fgf2均能够诱导细胞生长(n=3)。(b)将2ng/ml的纯化和内含肽切割的fgf2加入培养基中3天。在第0天和第3天拍摄相差显微照片。在fgf2处理3天后观察到pc12细胞中的延长的神经突生长(用箭头表示)。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
本发明提供了一种增强子序列,其特征在于,该增强子序列包括nf-κb结合位点的dna序列和creb结合位点的dna序列。
为了获得更好的增强效果,在一种优选的实施方式中,所述nf-κb结合位点的dna序列为seqidno.3所示:ggaaatccccggaaatcccc,或其互补序列;所述creb结合位点的dna序列为seqidno.4所示:tgcgtcaacactgctcaac,或其互补序列。
为了获得更好的增强效果,在另一种优选的实施方式中,所述增强子序列为47bp。
为了获得更好的增强效果,在一种优选的实施方式中,所述增强子序列为seqidno.5所示:ggaaatccccggaaatccccgtaaaatttgcgtcaacactgctcaac,或其互补序列。
本发明还提供了一种质粒载体,该质粒载体包括本发明所提供的增强子序列。
在一种优选的实施方式中,所述增强子序列位于质粒载体pcdna3.1(+)的nhei和hindiii限制性酶切位点之间。
为了获得更好的增强效果,在另一种优选的实施方式中,所述nf-κb结合位点的dna序列与nhei限制性酶切位点连接,creb结合位点的dna序列与hindiii限制性酶切位点连接。
本发明提供一种质粒载体pnc1的制备方法,其特征在于,该方法包括以下步骤:
a.合成含有nf-κb结合位点的dna序列和creb结合位点的dna序列的dna序列,其中,nf-κb结合位点的dna序列的末端带有nhei限制性酶切位点,creb结合位点的dna序列的末端带有hindiii限制性酶切位点;
b.用nhei和hindiii消化原始质粒载体,回收大片段;
c.采用连接酶连接步骤a获得的dna序列与步骤b获得的大片段;
d.将步骤c获得的连接产物转染细胞,筛选阳性克隆,提取质粒。
在一种优选的实施方式中,其中,所述nf-κb结合位点的dna序列为seqidno.3所示或其互补序列,所述creb结合位点的dna序列为seqidno.4所示或其互补序列。
在一种优选的实施方式中,其中,所述步骤a合成的dna序列为带有nhei和hindiii限制性酶切位点的seqidno.5所示序列或其互补序列。
为了获得更好的增强效果,在一种优选的实施方式中,其中,原始质粒载体为pcdna3.1(+)。
本发明的质粒载体pnc1可以用于多种蛋白质的表达,优选地,本发明还提供所述的质粒载体在表达fgf-2中的用途。
本发明还提供一种转化体,其特征在于,该转化体包括本发明提供的质粒载体pnc1。
所述转化体的宿主细胞没有特别限制,例如可以为hek293t、hela、c2c12等。但在一种优选的实施方式中,所采用的宿主细胞是hek293t。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
实施例
材料和方法
化学品、质粒和抗体,除非另有说明,否则所有化学品均购自sigma-aldrich(st.louis,mo)。
质粒载体pnc1的构建
如图1中的b1所示:
a.采用pcr(用geneartstrings基因合成服务(thermo-fisherscientific,waltham,ma))合成增强子序列,该增强子序列为含有nf-κb结合位点的dna序列和creb结合位点的dna序列的dna序列;
b.用nhei和hindiii消化质粒载体pcdna3.1(+);
c.用nhei和hindiii消化步骤a合成的dna序列;
d.采用连接酶连接步骤b获得的消化后的质粒载体pcdna3.1(+)与步骤c获得的消化后的dna序列。
通过sanger测序确认序列。
具体过程如下:
材料:pcdna3.1(+)购自美国thermoscientific公司,其载体图谱如图1a所示。限制性内切酶nhei和hindiii,以及t4dnaligase购自neb公司。质粒提取试剂盒和dna片段回收试剂盒购自thermoscientific公司。
增强子pcr扩增
增强子的模板通过geneartstringservice(thermosscientific)合成,其dna序列如seqidno.5所示:5’ggaaatccccggaaatccccgtaaaatttgcgtcaacactgctcaac。采用的正向和反向引物如表1所示。在ep管中依次加入以下反应物:0.5μl的模板,1x缓冲液,1μm正向引物,1μm反向引物,0.5μl聚合酶,4μldntp,加ddh2o至50μl反应体系中进行反应。
反应条件:95℃、5min;95℃、15s;55℃、10s;72℃、10s;35个循环;72℃延伸10min,4℃终止反应。
将pcr扩增产物在3%琼脂糖凝胶中电泳进行结果检测,用dna胶回收纯化试剂盒回收插入条带。获得59bp的片段,与预期大小一致。
将上述pcr回收产物连接至质粒载体pcdna3.1(+)上。
将上述pcr回收片段和质粒载体pcdna3.1(+)用nhei和hindiii酶切处理,然后用t4dna连接酶进行连接,连接产物转化至化学感受态细胞dh5a菌株中,涂布到氨苄抗性固体培养基平皿,挑取若干单克隆菌落,接种到氨苄抗性液体培养基中,恒温摇床37℃、250rpm振荡培养过夜。采用质粒提取试剂盒提取质粒,得到重组质粒载体pnc1。
重组质粒载体pnc1的鉴定
用nhei和hindiii酶切进行鉴定,酶切鉴定体系及反应条件如下表:
重组质粒dna5μl,10×缓冲液2μl,nhei0.5μl,hindiii0.5μl,ddh2o11μl
总体积20μl。37℃,15min。
经酶切鉴定,获得产物与预期分析大小一致。
选取酶切验证正确的阳性克隆进行测序分析,结果经finchtv解析及blast软件比对,以验证重组质粒载体的正确性。分析结果表明,增强子核苷酸序列完全正确,成功获得了重组质粒载体pnc1。
fgf2-pcdna3.1(+)构建体、fgf2-pnc1构建体和his-dnae-fgf2-pnc1构建体
没有前肽序列的fgf2基因(即fgf2基因的成熟功能片段)的dna序列(氨基酸143-288,pro_0000008933)被设计用于人类的密码子优化。
用geneartstrings基因合成服务(thermo-fisherscientific,waltham,ma)合成设计的fgf2基因,将合成的fgf2基因克隆到具有ecori和noti位点的pcdna3.1(+)和pnc1中,形成fgf2-pcdna3.1(+)构建体和fgf2-pnc1构建体,如图1a,1b2所示。通过sanger测序确认所有序列。
为了优化纯化,本发明使用6xhis标签以及使用融合了fgf2基因的点形念珠藻(nostocpunctiforme)pcc73102(npu)的dna聚合酶iii(dnae)内含肽来促进人成纤维细胞生长因子2(fgf2)的纯化。
用geneartstrings基因合成服务(thermo-fisherscientific,waltham,ma)合成设计的fgf2和6xhis-npudnae内含肽基因。通过重叠pcr将6xhis-npudnae内含肽与fgf2融合。
为了进一步提高fgf2的表达,使用geneartstrings基因合成服务合成含有nf-κb结合位点和creb结合位点的增强子序列,并克隆到具有nhei和hindiii位点的pcdna3.1(+)中(thermo-fisherscientific,waltham,ma))以形成表达质粒载体pnc1。将合成6xhis-dnae-fgf2)克隆到具有ecori和noti位点的pnc1中,形成his-dnae-fgf2-pnc1构建体,如图1c所示。通过sanger测序确认所有序列。用于蛋白质印迹的抗体:小鼠fgf-2(克隆c-2,santacruzbiotechnology,dallas,tx),小鼠抗β-肌动蛋白(sigma,st.louis,mo)。
fgf2-pcdna3.1(+)构建体的构建具体过程如下:
fgf2基因pcr扩增
fgf2的模板通过geneartstringservice(thermosscientific)合成,其dna序列如seqidno.1所示
ccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaagatccgaagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaagaacgaggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaagacggacggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggctagagtcaaacaattataacacctataggtcaagaaagtatacgagctggtacgttgcccttaagcggaccggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctcttcctccctatgagtgccaagtcttaataataa。采用的正向和反向引物如表1所示。在ep管中依次加入以下反应物:0.5μl的模板,1x缓冲液,1μm正向引物,1μm反向引物,0.5μl聚合酶,4μldntp,加ddh2o至50μl反应体系中进行反应。
反应条件:95℃、5min;95℃、15s;55℃、10s;72℃、30min;35个循环;72℃延伸10min,4℃终止反应。
将pcr扩增产物在1%琼脂糖凝胶中电泳进行结果检测,用dna胶回收纯化试剂盒回收插入条带。获得460bp的片段,与预期大小一致。
将上述pcr回收产物连接至质粒载体pcdna3.1(+)上。
将上述pcr回收片段和质粒载体pcdna3.1(+)用ecori和noti酶切处理,然后用t4dna连接酶进行连接,连接产物转化至化学感受态细胞dh5a菌株中,涂布到氨苄抗性固体培养基平皿,挑取若干单克隆菌落,接种到氨苄抗性液体培养基中,恒温摇床37℃、250rpm振荡培养过夜。采用质粒提取试剂盒提取质粒,得到fgf2-pcdna3.1(+)构建体。
fgf2-pcdna3.1(+)构建体的鉴定
用ecori和noti酶切进行鉴定,酶切鉴定体系及反应条件如下表:
fgf2-pcdna3.1(+)构建体5μl,10×缓冲液2μl,ecori0.5μl,noti0.5μl,ddh2o11μl总体积20μl。37℃,15min。
经酶切鉴定,获得产物与预期分析大小一致。
选取酶切验证正确的阳性克隆进行测序分析,结果经finchtv解析及blast软件比对,以验证fgf2-pcdna3.1(+)构建体的正确性。分析结果表明,fgf2核苷酸序列完全正确,成功获得了fgf2-pcdna3.1(+)构建体。
fgf2-pnc1构建体的构建具体过程如下:
fgf2基因pcr扩增
fgf2的模板通过geneartstringservice(thermosscientific)合成,其dna序列如seqidno.1所示:
ccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaagatccgaagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaagaacgaggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaagacggacggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggctagagtcaaacaattataacacctataggtcaagaaagtatacgagctggtacgttgcccttaagcggaccggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctcttcctccctatgagtgccaagtcttaataataa。采用的正向和反向引物如表1所示。在ep管中依次加入以下反应物:0.5μl的模板,1x缓冲液,1μm正向引物,1μm反向引物,0.5μl聚合酶,4μldntp,加ddh2o至50μl反应体系中进行反应。
反应条件:95℃、5min;95℃、15s;55℃、10s;72℃、30min;35个循环;72℃延伸10min,4℃终止反应。将pcr扩增产物在1%琼脂糖凝胶中电泳进行结果检测,用dna胶回收纯化试剂盒回收插入条带。获得460bp的片段,与预期大小一致。
将上述pcr回收产物连接至质粒载体pnc1上。
将上述pcr回收片段和质粒载体pnc1用ecori和noti酶切处理,然后用t4dna连接酶进行连接,连接产物转化至化学感受态细胞dh5a菌株中,涂布到氨苄抗性固体培养基平皿,挑取若干单克隆菌落,接种到氨苄抗性液体培养基中,恒温摇床37℃、250rpm振荡培养过夜。采用质粒提取试剂盒提取质粒,得到pnc1构建体。
fgf2-pnc1构建体的鉴定
用ecori和noti酶切进行鉴定,酶切鉴定体系及反应条件如下表:
fgf2-pnc1构建体5μl,10×缓冲液2μl,ecori0.5μl,noti0.5μl,ddh2o11μl总体积20μl。37℃,15min。经酶切鉴定,获得产物与预期分析大小一致。
选取酶切验证正确的阳性克隆进行测序分析,结果经finchtv解析及blast软件比对,以验证fgf2-pnc1构建体的正确性。分析结果表明,fgf2核苷酸序列完全正确,成功获得了fgf2-pnc1构建体。
his-dnae-fgf2-pnc1构建体的构建具体过程如下:
his-dnae-fgf2的融合pcr扩增
his-dnae-fgf2的模板通过geneartstringservice(thermosscientific)合成,其dna序列如seqidno.2所示:
catcatcaccatcaccacgccgagtactacgagaccgagatcctgaccgtggagtacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtgtacagcgtggacaacaacggcaacatctacacccagcccgtggcccagtggcacgacaggggcgagcaggaggtgttcgagtactgcctggaggacggcagcctgatcagggccaccaaggaccacaagttcatgaccgtggacggccagatgctgcccatcgacgagatcttcgagagggagctggacctgatgagggtggacaacctgcccaacgccgagtactacgagaccgagatcctgaccgtggagtacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtgtacagcgtggacaacaacggcaacatctacacccagcccgtggcccagtggcacgacaggggcgagcaggaggtgttcgagtactgcctggaggacggcagcctgatcagggccaccaaggaccacaagttcatgaccgtggacggccagatgctgcccatcgacgagatcttcgagagggagctggacctgatgagggtggacaacctgcccaacccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaagatccgaagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaagaacgaggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaagacggacggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggctagagtcaaacaattataacacctataggtcaagaaagtatacgagctggtacgttgcccttaagcggaccggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctcttcctccctatgagtgccaagtcttaataataa。以his-dnae正向引物和fgf2反向引物,采用的正向和反向引物如表2所示。在ep管中依次加入以下反应物:0.5μl的模板,1x缓冲液,1μm正向引物,1μm反向引物,0.5μl聚合酶,4μldntp,加ddh2o至50μl反应体系中进行反应。
反应条件:95℃、5min;95℃、15s;55℃、10s;72℃、30min;35个循环;72℃延伸10min,4℃终止反应。将pcr扩增产物在1%琼脂糖凝胶中电泳进行结果检测,用dna胶回收纯化试剂盒回收gfp条带。获得1097bp的片段,与预期大小一致。
将上述pcr回收产物连接至质粒载体his-dnae-fgf2上。
将上述pcr回收片段和质粒载体his-dnae-fgf2用ecori和noti酶切处理,然后用t4dna连接酶进行连接,连接产物转化至化学感受态细胞dh5a菌株中,涂布到氨苄抗性固体培养基平皿,挑取若干单克隆菌落,接种到氨苄抗性液体培养基中,恒温摇床37℃、250rpm振荡培养过夜。采用质粒提取试剂盒提取质粒,得到his-dnae-fgf2构建体。
his-dnae-fgf2构建体的鉴定
用ecori和noti酶切进行鉴定,酶切鉴定体系及反应条件如下表:
his-dnae-fgf2构建体5μl,10×缓冲液2μl,ecori0.5μl,noti0.5μl,ddh2o11μl总体积20μl。37℃,15min。
经酶切鉴定,获得产物与预期分析大小一致。
选取酶切验证正确的阳性克隆进行测序分析,结果经finchtv解析及blast软件比对,以验证his-dnae-fgf2构建体的正确性。分析结果表明,his-dnae-fgf2核苷酸序列完全正确,成功获得了his-dnae-fgf2构建体。
表1
pcr合成的含有nhei和hindiii限制性酶切位点增强子的dna为seqidno.11(即为含有nhei和hindiii限制性酶切位点seqidno.5):
gctagcggaaatccccggaaatccccgtaaaatttgcgtcaacactgctcaacaagctt。
pcr合成的含有ecori和noti限制性酶切位点的fgf2的dna序列为seqidno.12(即为含有ecori和noti限制性酶切位点seqidno.1):
gaattcccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaagatccgaagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaagaacgaggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaagacggacggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggctagagtcaaacaattataacacctataggtcaagaaagtatacgagctggtacgttgcccttaagcggaccggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctcttcctccctatgagtgccaagtcttaataataagcggccgc。
pcr合成的带有ecori和noti限制性酶切位点的his-dnae-fgf2的dna序列为seqidno.13(即为含有ecori和noti限制性酶切位点seqidno.2):
gaattcaccatgcatcatcaccatcaccacgccgagtactacgagaccgagatcctgaccgtggagtacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtgtacagcgtggacaacaacggcaacatctacacccagcccgtggcccagtggcacgacaggggcgagcaggaggtgttcgagtactgcctggaggacggcagcctgatcagggccaccaaggaccacaagttcatgaccgtggacggccagatgctgcccatcgacgagatcttcgagagggagctggacctgatgagggtggacaacctgcccaacgccgagtactacgagaccgagatcctgaccgtggagtacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtgtacagcgtggacaacaacggcaacatctacacccagcccgtggcccagtggcacgacaggggcgagcaggaggtgttcgagtactgcctggaggacggcagcctgatcagggccaccaaggaccacaagttcatgaccgtggacggccagatgctgcccatcgacgagatcttcgagagggagctggacctgatgagggtggacaacctgcccaacccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaagatccgaagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaagaacgaggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaagacggacggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggctagagtcaaacaattataacacctataggtcaagaaagtatacgagctggtacgttgcccttaagcggaccggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctcttcctccctatgagtgccaagtcttaataataagcggccgc。
细胞培养和转染
将hek293t和c2c12细胞维持在含有10体积%fbs和1体积%青霉素链霉素溶液(thermo-fisherscientific,waltham,ma)的dmem中,环境中温度为37℃,含有5%co2。将pc12细胞维持在含有10体积%hs,5体积%fbs和1体积%青霉素链霉素溶液的dmem中,环境中温度为37℃,含有5%co2。根据制造商的说明,用lipofectamine2000(thermo-fisherscientific,waltham,ma)将pcdna3.1-fgf2,pnc1-fgf2和pnc1-6xhis-dnae-fgf2构建体转染到hek293t中。
fgf2的纯化
收集培养基并以2,000g旋转10分钟以除去细胞碎片,并通过0.45μm过滤器过滤。然后将滤液通过用50mmtris-hcl(ph7.5)平衡的预装的肝素-琼脂糖柱(bioradlaboratories,hercules,ca)。用50mmtris-hcl(ph7.5),0.2mnacl彻底洗涤柱子。用0.3m至3m(4-5床体积用于梯度)的nacl梯度洗脱fgf2。洗脱后,使蛋白质通过用50mmtris-hcl(ph7.5)平衡的预装的sephadexg25柱,并用相同的缓冲液洗脱。
对于细胞内表达的fgf2,将细胞用冰冷的tbs洗涤三次,然后在裂解缓冲液(提供有completetm蛋白酶抑制剂混合物(completetmproteaseinhibitorcocktail)的tbs)中超声处理。然后在培养基中纯化fgf2。
为了获得内含肽切除的fgf2,通过预先包装的ni-nta柱纯化组氨酸标记的dnae-fgf2。洗涤后,将柱在50mmtris-hcl(ph6.2),10mmedta,200mmnacl中于22℃温育不同的持续时间,分别为0、1、2、4、6、10小时,以诱导dnae内含肽的c末端切除。
蛋白质分析
在15体积%tris-甘氨酸sds-page中分离蛋白质。凝胶用银染以获得纯化的蛋白质。在凝胶上,切下对应于fgf2的条带,洗涤并与1μg胰蛋白酶在50mmnh4hco3中于4℃温育过夜。通过与accelahple系统结合的ltqvelos线性离子阱质谱仪(ltqveloslineariontrapmassspectrometer)(thermofisherscienctific,sanjose)分析水解的样品。获得完整的ms扫描(300-2000m/z)并用mascot搜索引擎(matricscience,boston,ma)鉴定肽序列。对于细胞裂解物,将样品点样或转移到0.2μmnc膜(bioradlaboratories,hercules,ca)上,然后用抗体印迹(小鼠fgf-2(克隆c-2,santacruzbiotechnology,dallas,tx),小鼠抗β-肌动蛋白(sigma,st.louis,mo))。
fgf2的生物学测定
为了监测fgf2对细胞增殖的影响,如前所述进行mtt测定。简而言之,将c2c12细胞接种在96孔板上,并提供dmem+0.5体积%fbs和1ng/ml的商购fgf2,纯化的fgf2和内切肽切除的fgf2。通过加入mtt至终浓度1mg/ml测定细胞的活性,并在37℃下孵育6小时。然后用dmso替换培养基,并在微量板读数器中在540nm处测量吸光度。
为了检测fgf2的神经营养作用,将pc12细胞与提供有2ng/ml的fgf2的培养基一起培养3天。观察细胞的形态,并通过相差光学显微镜拍摄图像。
结果
构建表达人相同的外源fgf2的质粒
全长fgf2蛋白由288个氨基酸组成,其中氨基酸1-142被切割并除去以产生功能性fgf2。因此,氨基酸143-288的蛋白质序列经过密码子优化,用于智人(homosapiens)。为了探索蛋白质最大化表达的可行性,将nf-κb结合位点和creb结合位点增强子序列克隆到含有天然cmv立即早期启动子/增强子序列的pcdna3.1载体中。然后将合成的fgf2基因克隆到pcdna3.1和pnc1载体中(图1),用于比较两个克隆在哺乳动物细胞中的表达。实验表明pnc1载体大大提高了fgf2在哺乳动物细胞中的表达水平,如图2所示。新霉素抗性基因还通过g418硫酸盐提供选择标记。因此可以选择稳定转染的细胞用于纯化人相同的外源fgf2。
可溶性fgf2在hek293t细胞中的表达
选择hek293t细胞用于纯化,因为其易于转染。sv40大t抗原还提供细胞复制转染质粒的能力,其含有sv40复制起点。此外,cmv启动子是最强的启动子之一,并且在hek293t细胞中具有组成型活性。虽然hek293t细胞是贴壁细胞,但它适用于悬浮培养是通用的,因此有利于扩大fgf2的产生。将pcdna3.1-fgf2pnc1-fgf2和pnc1-6xhis-dnae-fgf2构建体瞬时转染至hek293t细胞并监测表达。实验表明:pnc1载体大大提高了fgf2的表达水平(图2a,2b)。fgf2的表达在转染后48小时达到峰值并且在较长时间点保持稳定状态。
使用细菌系统进行蛋白质制备和纯化的缺点之一是形成不溶性聚集体。为了测试在hek293t细胞中表达的fgf2的溶解度,从细胞裂解中收获沉淀级分。将不溶性级分在laemmili样品缓冲液中超声处理,然后在sds-page中电泳。裂解物的蛋白质印迹显示绝大部分fgf2是可溶的(图2c)。与fgf2标准相比,在pcdna3.1-fgf2中表达的fgf2约为20μg/ml,对于在pnc1-fgf2中表达的fgf2约为50μg/ml。
fgf2的纯化和蛋白质测序
为了使纯化的fgf2的产量最大化,用pnc1-fgf2转染hek293t细胞。转染后48小时,收获并裂解细胞以纯化细胞内表达的fgf2。通过肝素-琼脂糖亲和层析纯化fgf2,然后进行尺寸排阻层析。洗脱的fgf2在15%sds-page中电泳(图3a)。将样品冻干并在0.1xpbs中重构。为了检查fgf2的纯度,在电泳后进行银染色并且显示出非常高的纯度,在纯化的fgf2上方仅发现1个弱条带(图3b)。产量也令人满意。为了鉴定纯化的fgf2,切下sds-page上的条带,溶解并在胰蛋白酶消化后通过lc-ms进行分析。测序结果显示纯化的fgf2的一级结构与成熟的外源人fgf2相同(表2)。因此,我们预期纯化的fgf2表现出与人天然对应物相同的性质,活性和功能。
hek293t表达的fgf2具有生物学活性
已经显示fgf2在许多细胞类型中刺激细胞生长。在确认纯化的fgf2的一级结构后,我们用1ng/ml的纯化和商购fgf2处理培养的c2c12,pbs用作对照。然后通过mtt测定法测量培养细胞的活力(图4a)。两种fgf2样品都能够刺激c2c12细胞的增殖。纯化的fgf2显示出与商购fgf2相似甚至更高的生物活性,但差异不显着。fgf2还在神经元细胞系中表现出神经营养活性。因此,在pc12细胞中测试了纯化的fgf2的神经营养活性。在fgf2处理3天后,观察到延长的神经突生长,其在pbs对照组中不存在(图4b)。数据证实纯化的fgf2具有生物学活性,并具有与商购fgf2相似的活性。
在hek293t细胞中内含肽介导的纯化
将含有6x组氨酸标记的dnae-fgf2的重组蛋白质级联克隆到pnc1载体中(图1c),然后转染到hek293t细胞。收集培养基并以2,000g旋转10分钟以除去细胞碎片,并通过0.45μm过滤器过滤。通过ni-nta亲和层析纯化蛋白质。用裂解缓冲液(50mmtris-hclph6.2,10mmedta,200mmnacl)在ni-nta柱上进行fgf2切除,在22℃下进行不同的时间点。将珠子和上清液在2xlaemmili样品缓冲液中煮沸以洗脱珠子结合的蛋白质,然后进行sds-page(图5)。实验结果表明,内含肽的完全切割需要至少5小时的孵育。通过lc-ms测定切除的蛋白质的鉴定,发现切除的fgf2的一级序列与成熟的人fgf2相同(表3)。还测试了切除的fgf2对c2c12细胞的生物活性,并且内切蛋白切除的fgf2的性能也与纯化的fgf2相同(图6a,6b)。
表2
液相色谱-串联质谱法分析纯化的fgf2
a在胰蛋白酶部分消化纯化的fgf2后,通过mascot搜索引擎鉴定n-末端和c-末端序列
b肽的理论质荷比
c肽的实验质荷比
表3
用液相色谱-串联质谱法分析内含肽-切除的fgf2
a在胰蛋白酶部分消化纯化的fgf2后,通过mascot搜索引擎鉴定n-末端和c-末端序列
b肽的理论质荷比
c肽的实验质荷比
fgf2是一种非常有价值的蛋白质,具有广泛的效力,包括血管生成,神经发生和伤口愈合。阻碍fgf2在医学上使用的研究的原因之一是纯化和生物活性fgf2的高成本。使用hek293t细胞,我们成功表达并纯化了人源fgf2(图3)。银染的凝胶证实纯度非常令人满意。在c2c12和pc12细胞培养物中,纯化的fgf2表现出与市售fgf2相同的促有丝分裂或神经营养活性。这种简单的方案允许在哺乳动物系统中实验室规模生产人外源性fgf2,并且可以容易地按比例放大以进行大规模生产。
用于fgf2纯化的最广泛使用的宿主系统之一是细菌。然而,简单的原核生物缺乏必要的翻译后修饰,包括剪接,糖基化和二硫键,用于纯化的蛋白质的活性和溶解度。纯化蛋白质的天然折叠对其功能和溶解性至关重要。当在细菌系统中表达真核蛋白时经常发现蛋白质聚集或包涵体。包括在较低温度下诱导蛋白质或使蛋白质聚集体变性和复性的方法并不总是产生良好的产率。使用细菌系统的另一个问题是内毒素。脂多糖或内毒素通常存在于大肠杆菌中。内毒素的存在会引发人体免疫反应,并可能导致感染性休克。虽然有去除内毒素的商购试剂盒和方案,但内毒素的污染通常是不可避免的。然而,使用哺乳动物细胞,可以容易地克服上述问题。
使用哺乳动物细胞表达蛋白质,我们能够纯化功能性,正确折叠和修饰的蛋白质(表2,图3、图4)。因此,我们是第一次在哺乳动物细胞中成功纯化无标记的人成熟外源fgf2。
在本发明中,我们已经证明dnae是hek293t细胞中的快速切割内含肽。通过使用这种内含肽,我们已经证实切除的fgf2的一级结构(表3)和生物活性(图6)与其天然对应物相同。纯化的fgf2的医学应用将是广泛的。此外,质粒载体pnc1可用于表达多种有价值的蛋白质。
序列表
<110>钟树根
<120>通过内含子促进人类胚胎细胞因子-1在293t中表达的方法
<130>190064882
<160>13
<170>patentinversion3.5
<210>1
<211>447
<212>dna
<213>人工合成
<400>1
ccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaagatccg60
aagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggtcgggtg120
gacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaagaacga180
ggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaagacgga240
cggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggctagagtca300
aacaattataacacctataggtcaagaaagtatacgagctggtacgttgcccttaagcgg360
accggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctcttcctc420
cctatgagtgccaagtcttaataataa447
<210>2
<211>1077
<212>dna
<213>人工合成
<400>2
catcatcaccatcaccacgccgagtactacgagaccgagatcctgaccgtggagtacggc60
ctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtgtacagcgtggac120
aacaacggcaacatctacacccagcccgtggcccagtggcacgacaggggcgagcaggag180
gtgttcgagtactgcctggaggacggcagcctgatcagggccaccaaggaccacaagttc240
atgaccgtggacggccagatgctgcccatcgacgagatcttcgagagggagctggacctg300
atgagggtggacaacctgcccaacgccgagtactacgagaccgagatcctgaccgtggag360
tacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtgtacagc420
gtggacaacaacggcaacatctacacccagcccgtggcccagtggcacgacaggggcgag480
caggaggtgttcgagtactgcctggaggacggcagcctgatcagggccaccaaggaccac540
aagttcatgaccgtggacggccagatgctgcccatcgacgagatcttcgagagggagctg600
gacctgatgagggtggacaacctgcccaacccggctctcccagaggatggcggctcagga660
gcctttccaccaggacacttcaaagatccgaagaggctttactgcaagaatggtggattt720
ttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaatccgatccgcat780
ataaagctgcagctgcaagctgaagaacgaggggtggttagcataaagggcgtgtgtgct840
aataggtaccttgccatgaaagaagacggacggctcctcgcttctaagtgcgtgaccgac900
gagtgcttcttctttgagcggctagagtcaaacaattataacacctataggtcaagaaag960
tatacgagctggtacgttgcccttaagcggaccggccagtacaagcttggtagcaaaaca1020
ggccctggccagaaggctatcctcttcctccctatgagtgccaagtcttaataataa1077
<210>3
<211>20
<212>dna
<213>人工合成
<400>3
ggaaatccccggaaatcccc20
<210>4
<211>19
<212>dna
<213>人工合成
<400>4
tgcgtcaacactgctcaac19
<210>5
<211>47
<212>dna
<213>人工合成
<400>5
ggaaatccccggaaatccccgtaaaatttgcgtcaacactgctcaac47
<210>6
<211>30
<212>dna
<213>人工合成
<400>6
aaaagctagcggaaatccccggaaatcccc30
<210>7
<211>29
<212>dna
<213>人工合成
<400>7
aaaaaagcttgttgagcagtgttgacgca29
<210>8
<211>32
<212>dna
<213>人工合成
<400>8
aaagaattcccaccatgcatcatcaccaccat32
<210>9
<211>31
<212>dna
<213>人工合成
<400>9
aaagcggccgcttattattaagacttggcac31
<210>10
<211>31
<212>dna
<213>人工合成
<400>10
aaaagaattcgccaccatgccggctctccca31
<210>11
<211>59
<212>dna
<213>人工合成
<400>11
gctagcggaaatccccggaaatccccgtaaaatttgcgtcaacactgctcaacaagctt59
<210>12
<211>461
<212>dna
<213>人工合成
<400>12
gaattcccggctctcccagaggatggcggctcaggagcctttccaccaggacacttcaaa60
gatccgaagaggctttactgcaagaatggtggatttttcctccgcatccatccagacggt120
cgggtggacggcgtacgggagaaatccgatccgcatataaagctgcagctgcaagctgaa180
gaacgaggggtggttagcataaagggcgtgtgtgctaataggtaccttgccatgaaagaa240
gacggacggctcctcgcttctaagtgcgtgaccgacgagtgcttcttctttgagcggcta300
gagtcaaacaattataacacctataggtcaagaaagtatacgagctggtacgttgccctt360
aagcggaccggccagtacaagcttggtagcaaaacaggccctggccagaaggctatcctc420
ttcctccctatgagtgccaagtcttaataataagcggccgc461
<210>13
<211>1097
<212>dna
<213>人工合成
<400>13
gaattcaccatgcatcatcaccatcaccacgccgagtactacgagaccgagatcctgacc60
gtggagtacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgcaccgtg120
tacagcgtggacaacaacggcaacatctacacccagcccgtggcccagtggcacgacagg180
ggcgagcaggaggtgttcgagtactgcctggaggacggcagcctgatcagggccaccaag240
gaccacaagttcatgaccgtggacggccagatgctgcccatcgacgagatcttcgagagg300
gagctggacctgatgagggtggacaacctgcccaacgccgagtactacgagaccgagatc360
ctgaccgtggagtacggcctgctgcccatcggcaagatcgtggagaagaggatcgagtgc420
accgtgtacagcgtggacaacaacggcaacatctacacccagcccgtggcccagtggcac480
gacaggggcgagcaggaggtgttcgagtactgcctggaggacggcagcctgatcagggcc540
accaaggaccacaagttcatgaccgtggacggccagatgctgcccatcgacgagatcttc600
gagagggagctggacctgatgagggtggacaacctgcccaacccggctctcccagaggat660
ggcggctcaggagcctttccaccaggacacttcaaagatccgaagaggctttactgcaag720
aatggtggatttttcctccgcatccatccagacggtcgggtggacggcgtacgggagaaa780
tccgatccgcatataaagctgcagctgcaagctgaagaacgaggggtggttagcataaag840
ggcgtgtgtgctaataggtaccttgccatgaaagaagacggacggctcctcgcttctaag900
tgcgtgaccgacgagtgcttcttctttgagcggctagagtcaaacaattataacacctat960
aggtcaagaaagtatacgagctggtacgttgcccttaagcggaccggccagtacaagctt1020
ggtagcaaaacaggccctggccagaaggctatcctcttcctccctatgagtgccaagtct1080
taataataagcggccgc1097
- 还没有人留言评论。精彩留言会获得点赞!