用于开发胃癌诊断产品的分子标志物及应用的制作方法
本发明属于生物医药领域,涉及用于开发胃癌诊断产品的分子标志物及应用。
背景技术:
胃癌(Gastric Cancer,GC)是一种高死亡率的疾病,位居全球癌症相关死亡的第二位。2011年,有40多万患者在中国被诊断为胃癌,并且因胃癌死亡的患者约30万人(Di Gesualdo F,Capaccioli S,Lulli M.A pathophysiological view of thelong non-codingRNA world.Oncotarget.2014;5:10976-10996.)。环境因素和生活方式是造成这一现象的关键病因。尽管胃癌患者的治疗方案仍然在不断改进中,目前手术和全身化疗是主要的治疗方法(Kanat O,O'Neil BH.Metastatic gastric cancer treatment:a little slowbut worthy progress.Med Oncol.2013;30:464.)。然而,对胃癌发生发展的机制研究一直持续进行,但是确切分子机制仍然模糊。目前可以明确一些基因的改变与胃癌易感性有关,如癌基因,肿瘤抑制基因和生长的因素(Jemal A,Bray F,Center MM,Ferlay J,Ward E,Forman D.Global cancerstatistics.CA Cancer J Clin.2011;61:69-90.)。因此,为了提高胃癌早诊,改善预后,迫切需要在诊断中找到特定的分子标志物。
长链非编码RNA(long non-coding RNA,1ncRNA)是一类通常超过200nt的RNA,没有开放阅读框(Rinn JL,Chang HY.Genome regulation by long noncodingRNAs.Annu Rev Biochem.2012;81:145-166.)。根据其定位与转录方向,1ncRNA被分类为基因间1ncRNA(intergenic lncRNAs)和基因内1ncRNAs(intronic lncRNAs)(Di Gesualdo F,Capaccioli S,Lulli M.A pathophysiologicalview of the long non-coding RNA world.Oncotarget.2014;5:10976-10996.doi:10.18632/oncotarget.2770.)。在细胞核中,1ncRNA主要调控基因转录和mRNA拼接,同时他们影响核糖核酸细胞质中的稳定性和microRNA(miRNA)活性。胃癌相关1ncRNAs被广泛研究。LncRNAs参与多个肿瘤信号通路,如Notch,mTOR,NF-Kb,Wnt等。lncRNA参与调控细胞周期、增殖、凋亡、侵袭和转移。此外,越来越多研究表明1ncRNAs的异常表达对癌症的诊断具有临床意义。研究与胃腺癌相关的lncRNA,对于实现胃腺癌的早期诊断具有重要的意义,同时也成为目前研究的热点。
技术实现要素:
为了弥补现有技术的不足,本发明通过高通量测序技术结合及生物信息学的方法,筛选在胃腺癌中呈现差异表达的lncRNA分子标志物,通过检测分子标志物的表达水平,与参照水平进行对比,来判断受试者是否患有胃腺癌或者是否存在患有胃腺癌的风险。同时该分子标志物可以作为胃腺癌的特异性分子靶标,应用于胃腺癌的精准治疗。
为了实现上述目的,本发明采用如下技术方案:
本发明提供了检测RP11-199F11.2的试剂在制备诊断胃腺癌的产品中的应用。
进一步,当RP11-199F11.2表达上调时,受试者患有胃腺癌或者存在患有胃腺癌的风险。
进一步,所述产品包括通过核酸测序技术、核酸杂交技术、核酸扩增技术检测样本中RP11-199F11.2的表达水平的试剂。
核酸测序技术的示例性非限制性实例包括但不限于链终止子(Sanger)测序和染料终止子测序。本领域的普通技术人员将认识到,由于RNA在细胞中不太稳定并且在实验中更易受到核酸酶攻击,因此在测序前通常将RNA逆转录成DNA。
核酸测序技术的另一示例性非限制性实例包括下一代测序(深度测序/高通量测序),高通量测序技术是一种基于单分子簇的边合成边测序技术,基于专有的可逆终止化学反应原理。测序时将基因组的DNA的随机片段附着到光学透明的玻璃表面,这些DNA片段经过延伸和桥式扩增后,在玻璃表面形成数以亿计的簇,每个簇是具有数千份相同模板的单分子簇,然后利用带荧光基团的四种特殊脱氧核糖核苷酸,通过可逆性的边合成边测序技术对待测的模板DNA进行测序。
核酸杂交技术的示例性非限制性实例包括但不限于原位杂交(ISH)、微阵列和Southern或Northern印迹。原位杂交(ISH)是一种使用标记的互补DNA或RNA链作为探针以定位组织一部分或切片(原位)或者如果组织足够小则为整个组织(全组织包埋ISH)中的特异性DNA或RNA序列的杂交。DNA ISH可用于确定染色体的结构。RNA ISH用于测量和定位组织切片或全组织包埋内的mRNA和其他转录本(例如,ncRNA)。通常对样本细胞和组织进行处理以原位固定靶转录本,并增加探针的进入。探针在高温下与靶序列杂交,然后将多余的探针洗掉。分别使用放射自显影、荧光显微术或免疫组织化学,对组织中用放射、荧光或抗原标记的碱基标记的探针进行定位和定量。ISH也可使用两种或更多种通过放射性或其他非放射性标记物标记的探针,以同时检测两种或更多种转录本。
将Southern和Northern印迹分别用于检测特异性DNA或RNA序列。使从样本中提取的DNA或RNA断裂,在基质凝胶上通过电泳分离,然后转移到膜滤器上。使滤器结合的DNA或RNA与和所关注的序列互补的标记探针杂交。检测结合到滤器的杂交探针。该程序的一种变化形式是反向Northern印迹,其中固定到膜的底物核酸为分离的DNA片段的集合,而探针是从组织提取并进行了标记的RNA。
本发明可在检测前或同时地对核酸(例如,ncRNA)进行扩增。核酸扩增技术的示例性非限制性实例包括但不限于:聚合酶链式反应(PCR)、逆转录聚合酶链式反应(RT-PCR)、转录介导的扩增(TMA)、连接酶链式反应(LCR)、链置换扩增(SDA)和基于核酸序列的扩增(NASBA)。本领域的普通技术人员将认识到,某些扩增技术(例如,PCR)需要在扩增前将RNA逆转录成DNA(例如,RT-PCR),而其他扩增技术则直接扩增RNA(例如,TMA和NASBA)。
通常,PCR使用变性、引物对与相反链的退火以及引物延伸的多个循环,以指数方式增加靶核酸序列的拷贝数;RT-PCR则将逆转录酶(RT)用于从mRNA制备互补的DNA(cDNA),然后将cDNA通过PCR扩增以产生DNA的多个拷贝;TMA在基本上恒定的温度、离子强度和pH的条件下自身催化地合成靶核酸序列的多个拷贝,其中靶序列的多个RNA拷贝自身催化地生成另外的拷贝,TMA任选地包括使用阻断,部分、终止部分和其他修饰部分,以改善TMA过程的灵敏度和准确度;LCR使用与靶核酸的相邻区域杂交的两组互补DNA寡核苷酸。DNA寡核苷酸在热变性、杂交和连接的重复多个循环中通过DNA连接酶共价连接,以产生可检测的双链连接寡核苷酸产物;SDA使用以下步骤的多个循环:引物序列对与靶序列的相反链进行退火,在存在dNTPαS下进行引物延伸以产生双链半硫代磷酸化的(hemiphosphorothioated)引物延伸产物,半修饰的限制性内切酶识别位点进行的核酸内切酶介导的切刻,以及从切口3'端进行的聚合酶介导引的物延伸以置换现有链并产生供下一轮引物退火、切刻和链置换的链,从而引起产物的几何扩增。
进一步,所述试剂选自:特异性识别RP11-199F11.2的探针;或
特异性扩增RP11-199F11.2的引物;或
特异性分析RP11-199F11.2的芯片。
进一步,所述特异性扩增RP11-199F11.2的引物序列如SEQ ID NO.1~2所示。
本发明提供了一种体外检测RP11-199F11.2表达水平的产品,所述产品包括芯片、试剂盒。
进一步,所述芯片包括:固相载体,以及附着于其上的特异性识别RP11-199F11.2的探针。
作为非限制性实施例,所述固相载体可采用基因芯片领域的各种常用材料,例如但不限于尼龙膜,经活性基团(如醛基、氨基等)修饰的玻片或硅片、未修饰的玻片、塑料片等。
本发明芯片的制备可采用本领域已知的生物芯片的常规制造方法。例如,如果固相载体采用的是修饰玻片或硅片,探针的5’端含有氨基修饰的聚dT串,可将寡核苷酸探针配制成溶液,然后采用点样仪将其点在修饰玻片或硅片上,排列成预定的序列或阵列,然后通过放置过夜来固定,就可得到本发明的lncRNA芯片。
术语“探针”意欲包括在促进杂交的条件下与核酸或其补体中的靶标序列特异性杂交,从而允许检测靶标序列或其扩增的核酸的核酸寡聚体或适体。检测可以是直接(即由直接与靶标或扩增的序列杂交的探针产生)或间接(即由与连接探针和靶标或扩增的序列的中间分子结构杂交的探针产生)的。探针的“靶标”通常指扩增的核酸序列中与至少部分探针序列通过标准氢键或“碱基配对”特异性杂交的序列。“充分地互补”的序列允许探针序列与靶标序列稳定杂交,即使该两个序列不完全互补。探针可被标记或不标记。探针可通过具体DNA序列的分子克隆生产,也可被合成。本发明所属领域的技术人员可容易地确定可在本发明的背景下设计和使用的多种引物和探针。
“杂交”或“核酸杂交”或“杂交”通常指两个具有互补碱基序列,在适当条件下将形成热力学上稳定的双链结构的单链核酸分子的杂交。如本文所用的术语“杂交”可指在严格或非严格条件下的杂交。条件的设置在本领域技术人员的技术范围内,可根据本领域中说明的实验方案确定。
进一步,所述试剂盒包括:特异性扩增RP11-199F11.2的引物,特异性识别RP11-199F11.2的探针的探针或特异性分析RP11-199F11.2的芯片。
术语“引物”表示寡核苷酸,不管是在纯化的限制性消化物中天然存在还是合成产生,当置于诱导与核酸链互补的引物延伸产物合成的条件下,即在存在核苷酸和诱导剂如DNA聚合酶并在合适温度和pH下时它能作为合成起点。引物可以是单链或双链,并且必须足够长以在诱导剂存在下引发合成预期的延伸产物。引物的确切长度依赖于很多因素,其中包括温度、引物来源和使用方法。例如,对于诊断应用,依赖于靶序列的复杂性,寡核苷酸引物通常含有15-25个或更多核苷酸,尽管它可以含有更少核苷酸。参与确定引物适当长度的因素对于本领域技术人员容易知道。
进一步,所述试剂盒还包括选自下组的一种或多种物质:容器、使用说明书、阳性对照物、阴性对照物、缓冲剂、助剂或溶剂。
本发明提供了体外检测RP11-199F11.2表达水平的产品在制备诊断胃腺癌的工具中的应用。
本发明提供了RP11-199F11.2在制备治疗胃腺癌的药物组合物中的应用,所述药物组合物包括RP11-199F11.2的抑制剂。所述抑制剂为可以降低RP11-199F11.2水平的任何试剂。作为非限制性的实施例,包括:shRNA(小发夹RNA)、小干扰RNA(siRNA)、dsRNA、微小RNA、反义核酸,或能表达或形成所述shRNA、小干扰RNA、dsRNA、微小RNA、反义核酸的构建物。
在本发明中,RP11-199F11.2基因位于17号染色体上,包括RP11-199F11.2基因及其的同源物,突变,和同等型。该术语涵盖全长,未加工的RP11-199F11.2,以及源自细胞中加工的任何形式的RP11-199F11.2。该术语涵盖RP11-199F11.2的天然发生变体(例如剪接变体或等位变体)。一种代表性的RP11-199F11.2的序列如ENST00000571370.1所示。
术语“差异分子标志物表达”和“差异表达”可互换使用,指相对于其在正常对象中的表达,或相对于其在对特定治疗不同地应答的或具有不同的预后的患者中的表达,其表达在患有特定疾病的对象中被激活为较高或较低水平的分子标志物。该术语还包括其表达在相同的疾病的不同的阶段被激活为较高或较低水平的分子标志物。还要理解的是差异表达分子标志物可在核酸水平或蛋白水平上被激活或被抑制,或可经受选择性剪接以产生不同的多肽产物。这种差异可通过包括mRNA水平、微小RNA水平、lncRNA水平、反义转录物水平或蛋白表面表达、分泌或多肽的其他划分的多种改变来证实。差异分子标志物表达可包括两个或更多个基因之间或其基因产物之间的表达的比较;或两个或更多个基因之间或其基因产物之间的表达的比率的比较;或甚至相同基因的两个不同加工的产物的比较,其在正常对象和患病对象之间是不同的;或在相同疾病的不同阶段是不同的。差异表达包括例如在正常细胞和病态细胞之间或在经历不同的疾病事件或疾病阶段的细胞之间,在分子标志物中瞬时表达模式或细胞表达模式中的定量和定性的差异。
当分子标志物在个体中指示异常进程、疾病或其他病症或作为异常进程、疾病或其他病症的标志时,该分子标志物与在个体中指示正常进程、无疾病或其他病症或作为正常进程、无疾病或其他病症的标志的分子标志物的表达水平或值相比较,通常被描述为过表达的或低表达的。“上调”、“上调的”、“过表达”、“过表达的”可互换使用,指大于在健康或正常个体中通常检测到的分子标志物的值或水平(或值或水平的范围)的生物样品中的分子标志物的值或水平。该术语还可指大于在特定疾病的不同阶段可检测到的分子标志物的值或水平(或值或水平的范围)的生物样品中的分子标志物的值或水平。
“下调”、“下调的”、“低表达”、“低表达的”可互换使用,指小于在健康或正常个体中通常检测到的分子标志物的值或水平(或值或水平的范围)的生物样品中的分子标志物的值或水平。该术语还可指小于在特定疾病的不同阶段可检测到的分子标志物的值或水平(或值或水平的范围)的生物样品中的分子标志物的值或水平。
在本发明中,用于本文所描述的试剂、工具和/或说明书可于试剂盒中提供。例如,试剂盒可包含用于确定关于癌症患者的适当治疗的试剂、工具和说明书。所述试剂盒可包括用于从患者收集组织样品的试剂,例如通过活检来收集,和用于处理该组织的试剂。试剂盒还可包括用于进行分子标志物表达分析的一种或多种试剂,例如用于进行RT-PCR、qPCR、RNA印迹等以确定患者的样品中的分子标志物的表达水平的试剂。例如,所述试剂盒中可包括用于进行RT-PCR的引物,用于进行RNA印迹分析的探针。还可包括用于测定的适当的缓冲液。还可包括这些测定中的任何一种所需的检测试剂。
本文所表征的试剂盒还可包括描述如何进行用于测量分子标志物表达的测定的说明卡。说明卡还可包括如何确定参考组群的说明,包括如何确定参考组群中分子标志物的表达水平和如何集合表达数据以建立用于与试验患者相比较的参考。说明卡还可包括用于测定试验患者中的分子标志物表达和用于将该表达水平与参考组群中的表达相比较从而确定用于受试者的适当的化学治疗的说明。
试剂盒中包括的信息材料可以是与本文所描述的方法和/或用于本文所描述的方法的试剂的用途相关的描述的、指导的、销售的或其他的材料。例如,试剂盒的信息材料可包含联系信息,例如,物理地址、电子邮件地址、网站或电话号码,其中试剂盒的使用者可获得有关进行基因表达分析和解析结果的大量信息,尤其当应用于可能对特定治疗剂具有阳性应答的人类时。
附图说明
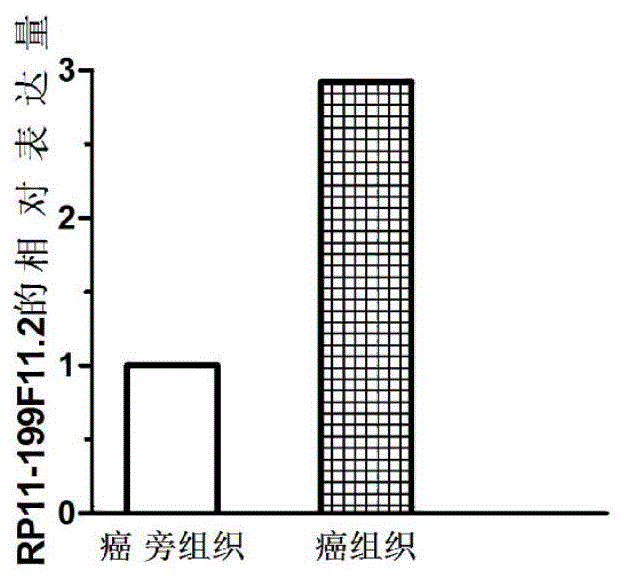
图1是利用QPCR检测RP11-199F11.2基因在胃腺癌组织中的表达情况图。
具体的实施方式
下面结合附图和实施例对本发明作进一步详细的说明。以下实施例仅用于说明本发明而不用于限制本发明的范围。实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
实施例1筛选与胃癌相关的基因标志物
1、样品收集
收集4例胃腺癌的癌组织及相对应的癌旁组织样本,进行高通量测序,所有患者术前未进行化疗、放疗以及内分泌治疗。
2、RNA样品的制备及质量分析
使用天根的动物组织总RNA提取试剂盒(目录号为DP431)进行总RNA的提取,步骤详见说明书。
1)匀浆处理
每10-20mg组织加300μl裂解液RL,用研磨杵将组织彻底研磨;随后向匀浆液中加入590μl RNase-Free ddH2O和10μl Proteinase K,混匀后56℃处理10-20min。
2)12,000rpm离心2-5min,取上清进行以下操作。
3)缓慢加入0.5倍上清体积无水乙醇,混匀,得到的溶液和沉淀一起转入吸附柱CR3中(吸附柱放在收集管中),12,000rpm离心30s,弃掉收集管中的废液,将吸附柱放回收集管中。
4)向吸附柱CR3中加入350μl去蛋白液RW1,12,000rpm离心30s,弃废液,将吸附柱放回收集管中。
5)向吸附柱CR3中央加入80μl的DNase I工作液,室温放置15min。
6)向吸附柱CR3中加入350μl去蛋白液RW1,12,000rpm离心30s,弃废液,将吸附柱放回收集管中。
7)向吸附柱CR3中加入500μl漂洗液RW,室温静置2min,12,000rpm离心30s,弃废液,将吸附柱CR3放回收集管中。
8)重复步骤7)。
9)12,000rpm离心2min,倒掉废液。将吸附柱CR3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液。
10)将吸附柱CR3转入一个新的RNase-Free离心管中,向吸附膜的中间部位悬空滴加30-100μl RNase-Free ddH2O,室温放置2min,12,000rpm离心2min,得到RNA溶液。
11)RNA的质量检测
用琼脂糖凝胶电泳检测RNA的完整性(电泳条件:胶浓度1.2%;0.5×TBE电泳缓冲液;150V,15min)检测完整性。28S rRNA是18S rRNA的两倍时,说明RNA的完整性较好。
用分光光度计检测RNA的浓度及纯度,OD260/OD280读数在1.8-2.1之间,RNA的质量较高。
3、cDNA文库的构建及测序
cDNA文库的构建及测序由华大基因完成,步骤如下:
1)总RNA DNase I消化:利用DNase I消化Total RNA样品中存在的DNA片段,磁珠纯化回收反应产物,最后溶解于DEPC水;
2)去除rRNA:取消化好的Total RNA样品,使用Epicentre的Ribo-Zero试剂盒去除rRNA,去除之后进行Agilent 2100检测,验证rRNA去除效果;
3)RNA打断:取上一步样品,加入打断Buffer,置于PCR仪中进行热打断,打断到140-160nt;
4)反转录一链的合成:向打断后的样品中加入适量引物,充分混匀后在Thermomixer适温反应一定时间,使之打开二级结构并与引物结合,再加入提前配制的一链合成反应体系Mix,在PCR仪上按相应程序合成一链cDNA;
5)反转录二链的合成:配制二链合成反应体系,在Thermomixer上适温反应一定时间,合成带有dUTP的二链cDNA,反应产物用磁珠进行纯化回收;
6)末端修复:配制末端修复反应体系,在Thermomixer中适温反应一定时间,在酶的作用下,对反转录得到的cDNA双链的粘性末端进行修复,末端修复产物用磁珠进行纯化回收,最后将样品溶于EB Solution;
7)cDNA 3’末端加“A”:配制加“A”反应体系,在Thermomixer中适温反应一定时间,在酶的作用下,使末端修复的产物cDNA的3’末端加上A碱基;
8)cDNA 5’adapter的连接:配制接头连接反应体系,在Thermomixer中适温反应一定时间,在酶的作用下,使接头与A碱基连接,产物用磁珠进行纯化回收;
9)UNG消化cDNA二链:配制UNG消化反应体系,通过UNG酶消化掉双链DNA中的二链,并用磁珠对产物进行纯化回收;
10)PCR反应及产物回收:配制PCR反应体系,选择适当的PCR反应程序,对上步骤得到的产物进行扩增,对PCR产物进行磁珠纯化回收,回收产物溶于EB溶液中,贴上标签。
11)文库质量检测:使用Agilent 2100Bioanalyzer和ABI StepOnePlusReal-Time PCR System对文库质量进行检测;
12)上机测序:检测合格的文库,加入NaOH变性成单链,按照预期上机数据量,稀释至一定的上机浓度。变性稀释后的文库加入到FlowCell内,与FlowCell上的接头杂交,在cBot上完成桥式PCR扩增,最后使用Illumina Hiseq x-ten平台进行测序。
4、生物信息学分析
1)用cutadapt对reads的5’和3’段进行trim,trim掉质量<20的碱基,并且删掉N大于10%的reads;
2)tophat比对到参考基因组上,参考基因组版本为GRCh37.p13;
3)cuffquant定量lncRNA的表达量并标准化输出;
4)cuffdiff包比较对照组跟疾病组lncRNA的表达差异。
5、结果
测序结果显示,与癌旁组织相比,RP11-199F11.2在胃腺癌患者中表达显著上调,提示RP11-199F11.2可能作为检测靶标应用于胃腺癌的早期诊断。
实施例2 QPCR测序验证RP11-199F11.2基因的差异表达
1、按照实施例1的收集方式收集的31例胃腺癌患者癌组织样本和癌旁组织样本对RP11-199F11.2基因差异表达进行大样本QPCR验证。
2、RNA提取
使用天根的动物组织总RNA提取试剂盒(目录号为DP431)进行总RNA的提取,具体步骤参见实施例1。
3、QPCR
根据RP11-199F11.2和GADPH的基因序列设计引物,引物序列如下:
RP11-199F11.2:
正向引物:5′-GGCTTCTGTCCTTCAATG-3′(SEQ ID NO.1)
反向引物:5′-CAAATGGGAAACAACTCAAA-3′(SEQ ID NO.2)
GAPDH:
正向引物:5′-AATCCCATCACCATCTTCCAG-3′(SEQ ID NO.3)
反向引物:5′-GAGCCCCAGCCTTCTCCAT-3′(SEQ ID NO.4)
使用天根公司的Quant一步法反转录-荧光定量试剂盒(SYBR Green)试剂盒(目录号:NG105)进行PCR反应,反应体系和反应条件如下面所示。在Thermal CyclerReal Time System扩增仪上进行PCR扩增,反应结束后确认Real Time PCR的扩增曲线和溶解曲线,ΔΔCT法进行相对定量。
50μl反应体系的配置
向EP管中加入下列试剂并混匀:2×Quant One Step RT-qPCR Mix(SYBR)25μl、Hotmaster Taq Polymerase 2.5U/μl 2.5μl、Quant RTase 0.4μl、正(反)向引物0.2μM、总RNA 50ng,加无核酶水至50μl。
反应条件
50℃30min,95℃2min;
(94℃20s,55℃20s,68℃20s)×40
4、结果
QPCR结果如图1所示,与对照相比,RP11-199F11.2在胃腺癌组织中表达上调,上调约2.9倍,差异具有统计学意义(P<0.05),同高通量测序结果一致,提示可通过检测RP11-199F11.2的水平判断受试者是否患有胃腺癌,与正常对照相比,当RP11-199F11.2的水平显著增加时,受试者患有胃腺癌或者存在患有胃腺癌的风险,通过RP11-199F11.2与胃腺癌之间的关系可以设计降低RP11-199F11.2水平的干扰RNA从而治疗胃腺癌。
上述实施例的说明只是用于理解本发明的方法及其核心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也将落入本发明权利要求的保护范围内。
序列表
<110> 徐州医科大学
<120> 用于开发胃癌诊断产品的分子标志物及应用
<160> 4
<170> SIPOSequenceListing 1.0
<210> 1
<211> 18
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 1
ggcttctgtc cttcaatg 18
<210> 2
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 2
caaatgggaa acaactcaaa 20
<210> 3
<211> 21
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 3
aatcccatca ccatcttcca g 21
<210> 4
<211> 19
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 4
gagccccagc cttctccat 19
- 还没有人留言评论。精彩留言会获得点赞!