一种可用于人原代细胞基因编辑的Cas9蛋白的制备方法与流程
一种可用于人原代细胞基因编辑的cas9蛋白的制备方法
技术领域
[0001]
本发明涉及蛋白纯化技术领域,更具体地,涉及一种可用于人原代细胞基因敲除的cas9蛋白的制备方法。
背景技术:
[0002]
crispr/cas是从细菌和古生菌对抗外来病毒或质粒的适应性免疫系统发展而来的新型核酸酶系统,包括三种不同的类型,其中typeⅱ型的crispr/cas系统的dna内切酶cas9只有一个亚基,结构最为简单,应用也最广泛。typeⅱ型的crispr/cas系统由导向序列sgrna和核酸酶cas9两种元件组成。sgrna的作用是识别基因组中核苷酸序列为5
’-
ngg-3’的pam基序,依据碱基互补配对原则与靶向序列结合,cas9在sgrna的引导下,特异性切割靶向序列,实现基因的插入和敲除。与zfn、talen相比,crispr/cas9系统具有操控性强、特异性高、用途广泛等优点。
[0003]
目前,crispr/cas系统可以用于在体内外进行特定位点的基因编辑,多篇文献报道中利用sgrna文库实现了细胞水平的基因功能性研究。将编码cas9和sgrna的质粒或rna转导至胚胎干细胞、受精卵或者胚胎,可快速制备果蝇、线虫、斑马鱼、小鼠、大鼠、食蟹猴和猪等多种基因敲除生物。另外,已有研究表明crispr/cas系统可在生殖干细胞和诱导多能干细胞(ipsc)中进行基因修复治疗遗传疾病。虽然目前对于crispr脱靶效应存在争议,但是随着基因治疗研究和实践的不断深入,其应用价值越来越引起人们的关注。
[0004]
crispr/cas系统有3种常见的应用模式,第一种是将cas9蛋白基因序列和sgrna构建到质粒载体上,然后将载体导入宿主细胞,利用宿主细胞自身表达cas9蛋白和sgrna。但是,受质粒转染效率的限制,使基因编辑过程无法快速高效地进行。第二种是将编码cas9蛋白的mrna和sgrna共同注射到宿主细胞中,但是由于mrna翻译延迟,也存在基因编辑效率低下或同一动物体内基因修饰不一致的问题。第三种也是目前比较理想的方式是将cas9以蛋白形式和sgrna共同导入宿主细胞,该过程不但降低了crispr/cas系统的导入难度,大大提高了基因编辑效率,同时cas9蛋白和sgrna以复合体的形式进入细胞或动物体内后立即行使功能,也缩短了作用时间。
[0005]
然而,目前对于cas9的研究多关注于使用crispr/cas平台技术进行基因编辑,然而实际应用中,获得纯化的cas9蛋白是后续研究和应用的关键技术手段。现有蛋白纯化手段仍比较繁琐,在蛋白表达纯化过程中引入的宿主细胞蛋白、宿主细胞dna和内毒素等相关杂质需要逐一去除,不易获得高纯度的蛋白满足动物乃至人体细胞试验的要求。尤其是内毒素,广泛存在于革兰氏阴性菌的细胞壁中,可能在大肠杆菌表达重组蛋白的过程中大量产生,内毒素作为一种热原,会导致生物体发生炎症发热反应,严重时可导致内毒素休克及散播性血管内凝血等。同时,纯化过程中使用的融合蛋白标签也会在后期的应用中带来一些不确定因素。因此急需一种简易且高效的cas9蛋白制备方法。
技术实现要素:
[0006]
本发明的目的在于提供一种简易且高效的cas9蛋白制备方法,在纯化过程中切除cas9蛋白的gst融合蛋白标签,并根据cas9蛋白带正电荷的特性,利用阴离子柱除去宿主细胞dna、内毒素及杂质蛋白等,得到高纯度和浓度的cas9蛋白并验证其体内外活性。
[0007]
为了实现上述目的,本发明提供以下的技术方案:
[0008]
一种去除内毒素的cas9蛋白的制备方法,其特征在于,包括如下步骤
[0009]
1)将cas9基因(type ii crispr rna-guided endonuclease cas9,streptococcus pyogenes;ncbi reference sequence:wp_010922251.1)克隆到pgex-4t-1载体上,插入gst标签切割位点,转化至原核表达菌株,诱导表达;
[0010]
2)破碎菌体并收集蛋白上清液进行gst标签结合柱纯化,并酶切gst标签;
[0011]
3)收集步骤2)的流穿液,调节其ph和离子强度,再使用阴离子交换柱去除杂质;
[0012]
4)收集步骤3)的cas9蛋白,超滤管浓缩得到高浓度去除内毒素的cas9蛋白。
[0013]
具体的,步骤1)所述插入的gst标签切割位点包括但不限于leu-glu-val-leu-phe-gln
▼-
gly-pro(seq id no.1,prescission protease的识别序列),leu-val-pro-arg
▼-
gly-ser(seq id no.2,thrombin的识别序列),ile-glu/asp-gly-arg
▼
(seq id no.3和seq id no.4,factor xa的识别序列)或asp-asp-asp-asp-lys
▼
(seq id no.5,enterokinase的识别序列)。
[0014]
其中使用factor xa和enterokinase酶切除后cas9蛋白上不会残留多余的的氨基酸残基,优选地插入factor xa或enterokinase切割位点。
[0015]
步骤1)所述cas9蛋白的诱导表达条件为:按照1:100的接种量,将表达的菌株接种于含氨苄抗性的lb培养基中,37℃培养至od600为0.8左右,摇床降温至16℃,继续培养半小时后在培养液中加入诱导物iptg至终浓度为0.5mmol/l,16℃,200rpm条件下,过夜培养。
[0016]
具体的,步骤2)所述破碎菌体方法为:使用140mm nacl,2.7mm kcl,10mm na2hpo4,1.8mm kh2po4组成的pbs(ph 8.0)对菌体进行裂解破碎。
[0017]
所述gst标签结合柱为gstsep glutathione agarose resin。
[0018]
所述gst标签结合柱纯化具体步骤为:
[0019]
①
对诱导后的菌体进行收集:离心,4000rpm 5min,弃去上清液;将菌体重悬到pbs缓冲液中超声波或高压破碎,破碎后的悬浮液低温高速离心取上清;
②
使用gst柱对蛋白进行纯化,用5倍树脂体积的pbs平衡gst柱子,3倍柱体积的pbs冲洗杂质,然后用gsh洗脱缓冲液(pbs+5mm gsh)对树脂进行蛋白液洗脱,测a280估计蛋白浓度。
[0020]
所述gst标签结合柱酶切具体步骤为:将收集到的gst-cas9蛋白于酶切缓冲液中透析,用合适的蛋白酶进行酶切,再利用gst蛋白纯化柱除去切下来的gst标签和没有被切割标签的gst-cas9蛋白,将流穿液收集。
[0021]
所述gst标签酶切所使用的酶包含但不限于prescission protease、thrombin、factor xa和enterokinase等。优选的,所述蛋白酶为factor xa或enterokinase。
[0022]
具体的,在进行步骤3)前还可以添加换液步骤,具体方法为:使用脱盐柱洗脱液对切除gst标签的cas9蛋白进行换液以进行下一步的阴离子柱的纯化。
[0023]
具体的,步骤3)使用阴离子交换柱去除杂质包括如下步骤:
[0024]
s1.活化、平衡树脂:将阴离子交换柱置于层析系统,调节系统流速为1ml/min,冲
洗5个柱体积的平衡液ph8.0,20mm tris+50mm nacl;
[0025]
s2.上样:将10ml的上样蛋白匀速推入上样环,流速为1ml/min;
[0026]
s3.样品收集:观察uv280的变化,当uv280成上升趋势时,收集流穿峰蛋白;
[0027]
s4.杂质及内毒素洗脱:用洗脱液20mm tris+1m nacl冲洗柱子,直到冲出杂质及内毒素,收集洗脱峰;
[0028]
s5.柱体再生保存:洗脱峰结束以后,对柱子进行cip,最后冲水至中性,再冲洗20%的乙醇保护柱子,取下柱子常温保存。
[0029]
具体的,步骤4)所述用超滤管浓缩cas9蛋白,蛋白浓度大于2mg/ml。
[0030]
可见,本申请提供了一种可用于基因编辑的cas9蛋白的制备方法,将cas9基因克隆到pgex-4t-1载体上,如有必要,在其5’端插入合适的gst标签切割位点,再转化至原核表达菌株,经过诱导表达后,破碎菌体并收集蛋白上清液进行纯化。在使用gst标签结合柱glutathione sehparose纯化过程中加酶切除gst标签,收集流穿液,调节其ph和离子强度。过阴离子柱,使杂质如大部分宿主细胞蛋白残余、宿主细胞dna残余、内毒素等结合在阴离子交换柱上,而带正电的cas9蛋白则不与阴离子层析柱或膜结合,从而达到蛋白纯化的目的,最后利用超滤管浓缩得到高浓度的cas9蛋白。
[0031]
其方法包括以下步骤:
[0032]
(1)为了使表达的cas9蛋白能在哺乳动物细胞内发挥剪切功能,我们在细菌的cas9cdna两端加入核定位序列。
[0033]
(2)cas9表达质粒构建:将带有核定位序列的cas9基因克隆到pgex-4t-1载体上,然后在n端插入合适的酶切位点,再转化至原核细胞表达cas9重组蛋白。插入的酶切位点包括但不限于leu-glu-val-leu-phe-gln
▼-
gly-pro(seq id no.1,prescission protease的识别序列),leu-val-pro-arg
▼-
gly-ser(seq id no.2,thrombin的识别序列),ile-glu/asp-gly-arg
▼
(seq id no.3和seq id no.4,factor xa的识别序列)或asp-asp-asp-asp-lys
▼
(seq id no.5,enterokinase的识别序列)中的任意一种。
[0034]
(3)优选的,所述酶切位点为ile-glu/asp-gly-arg
▼
(seq id no.3和seq id no.4,factor xa的识别序列)或asp-asp-asp-asp-lys
▼
(seq id no.5,enterokinase的识别序列)。
[0035]
(4)cas9蛋白表达的诱导条件为:按照1:100的接种量,将表达的菌株接种于lb培养基中,37℃培养8h至od600为0.8左右,加终浓度为0.5mm的iptg,于16℃过夜诱导表达。
[0036]
(5)gst柱纯化步骤:对诱导后的菌体进行收集:离心,4000rpm 5min,弃去上清液。将菌体重悬到pbs缓冲液中超声波或高压破碎。破碎后的悬浮液低温高速离心取上清。使用gst柱对蛋白进行纯化。用5倍树脂体积的pbs平衡gst柱子,3倍柱体积的pbs冲洗杂质,然后用gsh洗脱缓冲液(pbs+5mm gsh)对树脂进行蛋白液洗脱,测a280估计蛋白浓度。
[0037]
(6)gst标签酶切:将收集到的gst-cas9蛋白于酶切缓冲液中透析,用合适的蛋白酶进行酶切。再利用gst蛋白纯化柱除去切下来的gst标签和没有被切割标签的gst-cas9蛋白,将流穿液收集。
[0038]
(7)脱盐柱置换蛋白的缓冲液:使用脱盐柱对蛋白背景缓冲液进行置换,将蛋白置换到ph8.0,20mm tris+50mm nacl缓冲液中,层析系统流速2ml/min。观察系统a280变化,收集蛋白。
[0039]
(8)阴离子交换柱去除杂质蛋白和内毒素:使用阴离子交换柱去除目标蛋白以外的杂质蛋白及内毒素,平衡缓冲液为ph8.0,20mm tris+50mm nacl,洗脱缓冲液为ph8.0,20mm tris+1m nacl,系统流速为1ml/min。收集流穿峰及高盐洗脱峰,流穿峰为目的蛋白cas9,洗脱峰为杂质蛋白和内毒素。
[0040]
(9)浓缩:将去除杂质蛋白和内毒素后cas9蛋白,用0.22μm的滤膜过滤,再以10kda超滤管浓缩,得到浓度为2mg/ml的cas9蛋白,分装液氮速冻保存于-80℃。
[0041]
本发明所述方法纯化得到的cas9蛋白经体外切割试验证明具有体外活性,可特异切割dna序列;同时经过改造的cas9蛋白n端和c端带有核定位序列,经电转化进入人原代细胞后可对特定基因进行敲除,证明纯化的cas9蛋白具有体内活性,可用于原代细胞的基因编辑。
[0042]
由于动物体内基因编辑和人原代细胞的特殊性,需要高纯度的试验材料且不含外源杂质,本发明制备的cas9蛋白去除了融合标签,减少其对蛋白结构和功能的影响,同时在纯化过程中利用大多数杂质蛋白和内毒素带负电而cas9蛋白带正电的特性,利用阴离子柱去除杂质蛋白和内毒素,经检验,cas9蛋白中的宿主细胞蛋白、宿主细胞dna、内毒素含量降到临床试验允许的水平(具体检验标准为《中国药典》2015年版第三部通则3412,《中国药典》2015年版第三部通则3407,《中国药典》2015年版第三部通则1143)。另外,根据本发明所述制备方法得到的cas9蛋白的检验标准及体内外活性检测方法也在本发明的保护范围之内。
[0043]
与现有技术相比本发明的有益效果为:
[0044]
1.cas9蛋白纯化过程中切除gst融合蛋白标签,减少标签对蛋白结构和功能的影响,降低蛋白免疫原性;其中使用factor xa和enterokinase酶切除后cas9蛋白上不会残留多余的的氨基酸残基,优选地插入factor xa或enterokinase切割位点。
[0045]
2.在纯化过程中利用cas9蛋白和内毒素等杂质带电性的不同,利用阴离子柱一步去除杂质蛋白和内毒素,简化杂质去除步骤,提高蛋白回收率,降低生产成本;
[0046]
3.本发明说述方法纯化得到的cas9蛋白经体外切割试验证明具有体外活性,可特异切割dna序列;经电转化进入人原代细胞后可对特定基因进行敲除,证明纯化的cas9蛋白具有体内活性,可用于原代细胞的基因编辑。
附图说明
[0047]
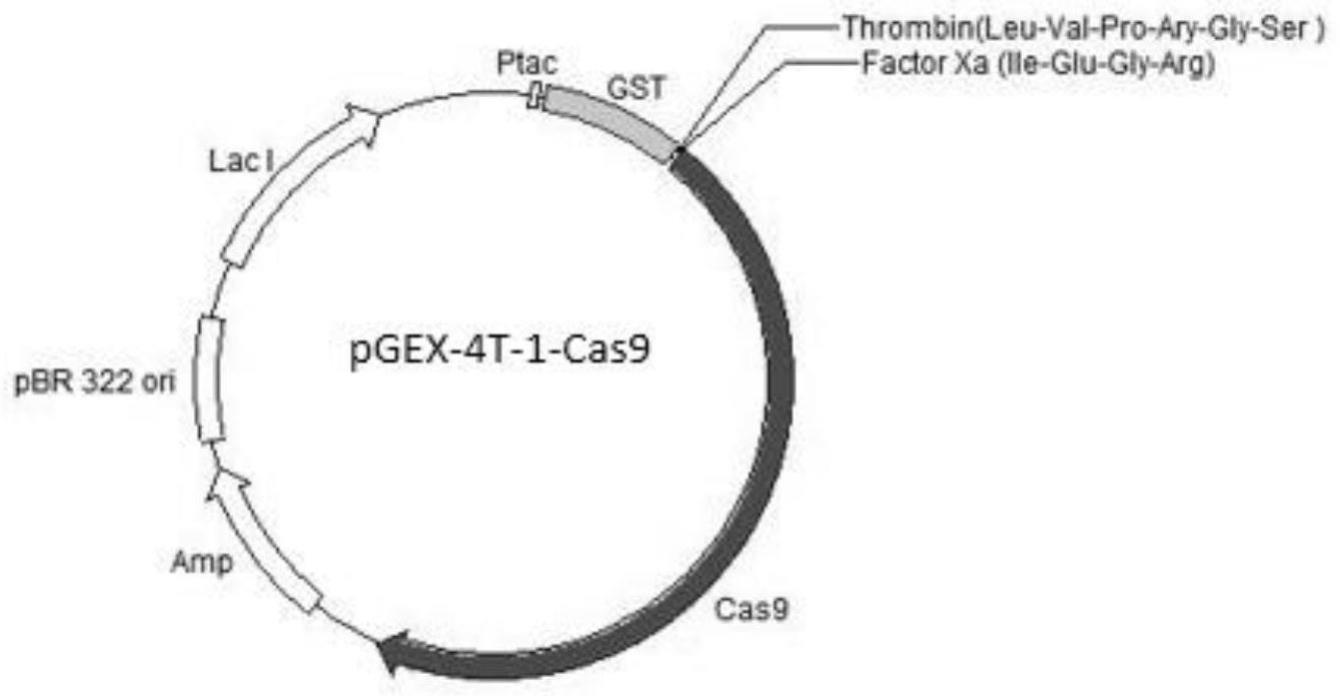
图1为重组质粒pgex-4t-1-cas9载体图谱。
[0048]
图2为酶切gst标签前后蛋白胶图验证,其中,
[0049]
m:marker;1:gst-cas9蛋白;2:酶切的cas9及gst标签混合物;3:去除gst标签的cas9蛋白。
[0050]
图3为cas9蛋白过脱盐柱前后蛋白胶图,其中,
[0051]
m:marker;1:脱盐柱上样蛋白;2-3:过脱盐柱后收集cas9蛋白。
[0052]
图4为cas9蛋白过阴离子交换柱前后蛋白胶图,其中,
[0053]
m:marker;1:阴离子上样蛋白;2:阴离子流穿峰(cas9蛋白);3:阴离子洗脱峰。
[0054]
图5为cas9蛋白体外活性实验,其中;
[0055]
m:marker;1:阴性对照(无cas9蛋白);2:阳性对照(市售的0.5ug的cas9蛋白);3、
4:实验组(0.5ug根据该方法纯化的cas9蛋白)。
[0056]
图6为体内活性试验,其中;
[0057]
nc:没有电转的样品;1-5:进行电转的样品。
具体实施方式
[0058]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
[0059]
除非特别说明,以下实施例所用的试剂和材料均为市购。
[0060]
实施例1:构建重组质粒pgex-4t-1-cas9及cas9蛋白的原核表达
[0061]
1.1、构建重组质粒pgex-4t-1-cas9载体
[0062]
在载体pgex-4t-1的gst序列的c端连接factor xa酶切位点,并在酶切位点的c端连接入带有核定位序列的cas9片段。
[0063]
质粒图谱见图1。其中thrombin酶识别位点的碱基序列为seq id no.6:ctggttccgcgtggatcc(seq id no.2:leu-val-pro-arg-gly-ser);factor xa识别位点的碱基序列为seq id no.7:atcgaaggtcgt(seq id no.3:ile-glu-gly-arg)。由于factor xa的切割位点在识别序列ile-glu-gly-arg
▼
的最后一位,所以经factor xa酶切后,cas9蛋白不含任何多余的氨基酸残基。
[0064]
1.2、重组质粒pgex-4t-1-cas9的原核表达
[0065]
1).将质粒pgex-4t-1-cas9转化至de3表达菌株中;
[0066]
2).挑取重组菌的单克隆至25ml lb培养基中37℃培养过夜,按照1:100的接种量,将过夜菌株接至1l的lb培养基中37℃培养至od600为0.8左右,16℃继续培养30min,加iptg至终浓度为0.5mm,16℃过夜培养;
[0067]
3).收集菌体:4000rpm离心5min,弃去上清液;
[0068]
4).破碎细胞:将菌体悬浮到一定量的pbs缓冲液中,然后超声波破碎;
[0069]
5).蛋白提取液的收集:将经超声破碎后的菌体混悬液12000g离心20min,取上清,即蛋白提取液。
[0070]
实施例2:cas9蛋白的纯化
[0071]
2.1、gst-cas9蛋白的纯化
[0072]
1).gst柱处理:取出gst柱底帽,滴出多余液体,柱子顶部朝上直立固定好,蒸馏水洗涤一次,再用5倍树脂体积的pbs平衡柱子;
[0073]
2).介质与蛋白结合:从柱子顶部加入蛋白提取液,直到加完为止。收集流穿液,洗涤:用3倍胶柱体积的pbs洗gst柱;
[0074]
3).样品收集:gsh洗脱缓冲液加入树脂内,收集洗脱液。测a280,估算洗脱的gst-cas9蛋白量;
[0075]
4).柱子洗涤:加入3倍树脂体积的pbs洗涤柱子,再加入10倍柱体积的20%的乙醇进行保存。
[0076]
2.2、酶切gst-cas9蛋白
[0077]
将收集到的gst-cas9蛋白透析到透析液(ph 8.0,20mm tris-hcl,100mm nacl和2mm cacl2)中,4℃过夜。次日用factor xa,按照产品说明书所述条件进行酶切。
[0078]
2.3、gst柱洗脱方法
[0079]
将切除标签的的cas9蛋白与标签的混合,利用gst蛋白纯化柱除去切下来的gst标签和没有被切割标签的gst-cas9蛋白,将流穿液收集。标签切除效果如附图2。
[0080]
2.4、脱盐柱置换cas9蛋白的缓冲液
[0081]
1).活化、平衡树脂:调节系统流速为2ml/min,调整系统的基线为0,冲洗5个柱体积的平衡液ph 8.0,20mm tris+50mm nacl;
[0082]
2).上样:暂停层析系统,使用5ml的注射器吸取3ml的上样蛋白并匀速推入上样环,继续使用2ml/min启动层析系统,观察uv280与uv230的变化;
[0083]
3).收集蛋白:收集蛋白峰,并留样跑胶;
[0084]
4).柱体再生保存:样品收集结束以后,使用5个柱体积平衡缓冲液ph 8.0,20mm tris+50mm nacl平衡柱子。并用20%的乙醇保护柱子,取下柱子常温保存。
[0085]
脱盐柱过柱前后蛋白情况如附图3。
[0086]
2.5、去除cas9蛋白中的杂质和内毒素
[0087]
1).活化、平衡树脂:调节系统流速为1ml/min,调整系统的基线为0,冲洗5个柱体积的平衡液ph 8.0,20mm tris+50mm nacl;
[0088]
2).上样:暂停层析系统,使用10ml的注射器吸取10ml的上样蛋白并匀速推入上样环,继续使用1ml/min的流速启动层析系统;
[0089]
3).样品收集:观察uv280与uv230的变化,收集流穿峰蛋白;
[0090]
4).杂质及内毒素洗脱:待收集流穿峰蛋白后,用洗脱液ph 8.0,20mm tris+1m nacl冲洗柱子,直到冲出杂质及内毒素,收集洗脱峰进行跑胶;
[0091]
5).柱体再生保存:洗脱峰结束以后,使用5个柱体积平衡缓冲液ph 8.0,20mm tris+50mm nacl平衡柱子,并用20%的乙醇保护柱子,取下柱子常温保存。
[0092]
2.6、浓缩:将去除杂质蛋白和内毒素后的cas9蛋白,用0.22μm的滤膜过滤,再以10kda超滤管浓缩,得到浓度为2mg/ml的cas9蛋白,分装液氮速冻保存于-80℃。
[0093]
2.7、cas9蛋白纯化结果检测
[0094]
cas9蛋白的目标条带约162kd(包含n段和c端的核定位序列),如附图4,经过上述纯化步骤可以看出,最终得到的cas9蛋白的条带清晰,用软件扫描灰度发现,蛋白的纯度为95%以上。
[0095]
实施例3:cas9蛋白的活性检测
[0096]
3.1、cas9蛋白体外活性检测
[0097]
cas9蛋白与sgrna共同作用可以对目标dna序列进行体外切割。我们选择tp53基因作为检验cas9蛋白活性的底物,设计针对tp53基因的sgrna(seq id no.8):gagcgctgctcagatagcgatgg,并进行体外转录,将cas9蛋白与sgrna复合物加入经pcr扩增(f:ccaactctctctagctcgc(seq id no.9);r:acggggtttcaccgttagcc(seq id no.10))的tp53基因片段中,电泳显示该片段被切为两个片段片段大小分别为270bp和530bp,从而确定cas9蛋白具有体外活性。
[0098]
检测体外活性的具体方法为:将100ng靶dna、100ng的sgrna和0.5ug cas9蛋白加入10ul作用体系中,37℃反应1h,65℃变性10min,使用0.8%的琼脂糖凝胶对切割效果进行分析。只加入sgrna时,靶dna不能被切开;将cas9蛋白与sgrna复合物加入经pcr扩增的tp53
片段中,750bp的靶dna片段被切割成270bp和530bp的片段,由此可知纯化后得到的cas9蛋白具有很好的体外活性,如图5所示。
[0099]
3.2、cas9蛋白体内活性检测
[0100]
把cas9蛋白(16ug)和sgrna(8ug)按比例混合至10ul体系,37℃孵育10min;将分选激活的人原代t细胞吹吸混匀,在室温下1000rpm离心5min,使细胞沉淀,去掉上清培养基,用电转缓冲液r重悬细胞,使用电转系统进行电转。电转后72h对t细胞tp53蛋白进行western blot检测,通过计算每个样品目的基因与内参基因的灰度比值,从而计算出cas9蛋白-grna复合物对tp53的敲除效果。通过附图6可知,纯化的cas9蛋白具有较高的体内活性。
[0101]
实施例4:cas9蛋白的内毒素检测
[0102]
使用鲎试剂内毒素检测试剂盒检测cas9蛋白的内毒素含量,基本原理为:鲎试剂内毒素检测试剂盒中含有c因子、b因子、凝固酶原、凝固蛋白原等,在适宜的条件下(温度,ph值及无干扰物质),细菌内毒素激活c因子,引起一系列酶促反应,使鲎试剂产生凝集反应形成凝胶。如果待检产品为阴性,则无法形成凝胶。
[0103]
具体检测方法为:将cas9蛋白浓度代入公式(0.25eu/ml*0.2ml)/(2.1μg/μl*0.5eu/μg)中计算蛋白的加入量,取相应量的cas9蛋白用无菌注射用水稀释到待测体积,同时,取同等体积的无菌注射用水转入鲎试剂管中作为阴性对照;将鲎试剂检测管架在悬浮漂中,37℃水浴锅中反应1h,观察是否凝结;若凝结则为不合格,若仍然为液体状则为合格。
[0104]
以上详细描述了本发明的优选实施例,但是,本发明并不限于上述实施例中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0105]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
[0106]
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1