一种丙泊酚磷酰胺衍生物、其制备方法及其在医药上的用途与流程
[0001]
本发明涉及一种丙泊酚磷酰胺衍生物及其中间体和制备方法,以及在制备药物中的用途。
背景技术:
[0002]
丙泊酚(propofol),化学名称为2,6-二异丙基苯酚,是目前临床上广泛应用的烷基酸类短效静脉全身麻醉药品,在国内外静脉麻醉药物市场处于领先和主导地位。静脉注射后迅速分布于全身,40秒钟内可产生睡眠状态,进入麻醉迅速、平稳。在临床使用上,同其他麻醉药物相比,丙泊酚起效快速,苏醒快速,且药代动力学和药效动力学完全可控、适用症广泛、禁忌症少,普通病人和高危病人均可安全使用。
[0003]
丙泊酚的酚羟基容易发生氧化反应,产生降解杂质,稳定性差。因异丙基的位阻影响,丙泊酚水溶性很差,目前普遍将丙泊酚配制成脂肪乳剂用于静脉注射麻醉,会产生局部疼痛,较大的油滴有引起栓塞的风险;脂肪乳会加重脂质代谢负荷,易污染导致细菌感染及过敏反应等风险。
[0004]
根据前药的原理,将丙泊酚的羟基进行前体改造,以期改善其溶解度、稳定性,以达到改善药物的理化性质、改善药物在体内的吸收、分布、代谢与排泄过程、提高口服生物利用度、提高药物对靶部位作用的选择性、降低药物的毒副作用、延长作用时间等的技术效果。这类药物通过口服进入体内后,可按照设计要求以一定的水解速度释放出2,6-二异丙基苯酚衍生物,产生麻醉、镇静和催眠效应,从而克服丙泊酚水溶性差和稳定性差的缺点。
技术实现要素:
[0005]
本发明通过磷酰胺基团改变丙泊酚的物理化学性质,如改变丙泊酚原有的性状、稳定性、脂溶性等,进而改变体内跨膜吸收、分布及代谢行为。磷酰胺修饰的药物进入体内后,在体内水解酶作用下水解释放出丙泊酚。通过控制磷酰胺药物水解速率可以延长药物体内的存在时间,也可以通过水解酶的分布等特点达到提高药物对靶部位的特异性作用给药的目的。
[0006]
本发明目的在于提供表1所示化合物、其立体异构体或其药学上可接受的盐,
[0007]
表1
[0008]
[0009][0010]
或其立体异构体或其可药用的盐。
[0011]
本发明提供一种化合物(1)的结晶固态形式,其特征在于使用cu-ka辐射,得到以2θ角度和晶体间距(d值)表示的x-射线粉末衍射图谱,其中在7.203,8.715,8.918,10.897,14.409,16.757,19.805,21.669,23.188,24.432,25.696,26.642,29.026,30.272有特征峰。
[0012]
本发明提供一种化合物(1)的结晶固态形式,其特征在于所述的结晶在差示扫描热分析中在78.80
±
3℃有一熔融吸热峰,在272.71
±
3℃有一分解吸热峰。
[0013]
本发明提供一种化合物(2)的结晶固态形式,其特征在于使用cu-ka辐射,得到以2θ角度和晶体间距(d值)表示的x-射线粉末衍射图谱,其中在7.089,7.365,8.823,9.111,11.120,13.150,14.720,16.718,16.974,17.116,20.285,20.803,22.227,23.755,24.336,25.002,26.296,27.338,29.325,29.735,33.837,34.665有特征峰。
[0014]
本发明提供一种制备根据权利要求1所述的化合物(1)或化合物(2)的晶体的方法,该方法包括:
[0015]
(1)将非对映异构体化合物(1e)或(2e)溶解于适量的有机溶剂或该有机溶剂的水溶液中,低温下放置,析出单一构型的化合物(1)或化合物(2)的晶体;
[0016]
(2)过滤结晶并洗涤,干燥。
[0017]
本发明提供一种制备根据权利要求1所述的化合物(1)或化合物(2)的晶体的方法,其特征在于,在步骤(1)中低温下放置的温度选自
–
60℃~25℃,优选
–
40℃~0℃,最优选
–
20℃。
[0018]
本发明提供一种制备根据权利要求1所述的化合物(1)或化合物(2)的晶体的方法,其特征在于,在步骤(1)中将消旋体化合物(1e)或(2e)溶解于适量的有机溶剂或该有机溶剂的水溶液中,低温下放置的时间选自0~21天,优选为7天。
[0019]
本发明提供一种制备根据权利要求1所述的化合物(1)或化合物(2)的晶体的方法,其特征在于,在步骤(1)中所述的有机溶剂选自石油醚、正己烷、环己烷、甲基环己烷、正戊烷、环戊烷、正庚烷、环庚烷、乙酸乙酯、二氯甲烷、丙酮、乙腈、n,n-二甲基甲酰胺、甲醇或乙醇的单一或不同比例的混合溶剂,优选选自正己烷、环己烷、甲基环己烷、正戊烷、环戊烷、正庚烷或乙酸乙酯,最优选为正庚烷。
[0020]
本发明涉及一种药物组合物,所述药物组合物含有治疗有效剂量的通式(i)所示的化合物、其立体异构体或其药学上可接受的盐,以及药学上可接受的载体和赋形剂。
[0021]
本发明涉及一种通式(i)所示的的化合物、其立体异构体或其药学上可接受的盐,或其组合物,在制备治疗和/或预防神经精神系统相关疾病的药物中的用途,其中所述的神经精神系统相关疾病选自焦虑、抑郁、失眠、恶心、呕吐、偏头痛、精神分裂、惊厥和癫痫。
[0022]
本发明涉及一种通式(i)所示的的化合物、其立体异构体或其药学上可接受的盐,或其药物组合物,在制备药物中的用途,所述用途包含:诱导和维持动物或者人类的麻醉、
镇静、器官组织缺氧缺血保护、促进动物或者人类的镇静催眠、治疗和/或预防焦虑、抑郁、失眠、恶心、呕吐、偏头痛、精神分裂、惊厥和癫痫药物中的用途,优选在制备诱导和维持动物或者人类的麻醉药物中的用途。
[0023]
本发明涉及一种通式(i)所示的的化合物、其立体异构体或其药学上可接受的盐,或其药物组合物,在制备用于提高药物口服生物利用度、延长母体药物的药效学半衰期、降低给药剂量和频率或延长半衰期的药物中的用途。
[0024]
发明的详细说明
[0025]
除非有相反的陈述,在说明书和权利要求书中使用的术语具有下述含义。
[0026]“药物组合物”表示一种或多种文本所述化合物或其生理学/药学上可接受的盐与其他组成成分的混合物,其中其它组分包含生理学/药学上可接受的载体和赋形剂。
[0027]“载体”指的是不会对生物体产生明显刺激且不会消除所给予化合物的生物活性和特性的载体或稀释剂。
[0028]“赋形剂”指的是加入到药物组合物中以进一步依赖于化合物给药的惰性物质。赋形剂的实例包括但不限于碳酸钙、磷酸钙、各种糖和不同类型的淀粉、纤维素衍生物(包括微晶纤维素)、明胶、植物油、聚乙二醇类、稀释剂、成粒剂、润滑剂、粘合剂、崩解剂等。
[0029]“前药”是指可以在生理条件下或通过溶剂解转化为具有生物活性的本发明化合物的化合物。本发明的前药通过修饰本发明化合物中的功能基团来制备,该修饰可以通过常规的操作或者在体内被除去,而得到母体化合物。
[0030]“立体异构体”是指由分子中原子在空间上排列方式不同所产生的异构体,包括顺反异构体、对映异构体和构象异构体。
[0031]“有效剂量”指引起组织、系统或受试者生理或医学反应的化合物的量,此量是所寻求的,包括在受治疗者身上施用时足以预防受治疗的疾患或病症的一种或几种症状发生或使其减轻至某种程度的化合物的量。
[0032]
本发明所述的“x射线粉末衍射图谱”是指根据布拉格公式2d sinθ=nλ(式中,λ为x射线的波长,衍射的级数n为任何正整数,一般取一级衍射峰,n=1),当x射线以掠角θ(入射角的余角,又称为布拉格角)入射到晶体或部分晶体样品的某一个具有d点阵平面间距的原子面上时,就能满足布拉格方程,从而测得了这组x射线粉末衍射图。
[0033]
本发明所述的“2θ或2θ角度”是指衍射角,θ为布拉格角,单位为
°
或度,2θ的误差范围为
±
0.1~
±
0.5,优选为
±
0.1~
±
0.3,更优选为
±
0.2。
[0034]
本发明所述的“晶面间距或晶面间距(d值)”是指空间点阵选择3个不相平行的连接相邻两个点阵点的单位矢量a,b,c,它们将点阵划分成并置的平行六面体单位,称为晶面间距。空间点阵按照确定的平行六面体单位连线划分,获得一套直线网格,称为空间各自或晶格。点阵和晶格是分别用几何的点和线反映晶体结构的周期性,不同的晶面,其面间距(即相邻的两个平行晶面之间的距离)各不相同;单位为或埃。
[0035]
本发明所述的“x射线单晶衍射图谱”仅用于鉴定样品绝对构型。
[0036]
本发明所述的“差示扫描量热分析或dsc”是指在样品升温或恒温过程中,测量样品与参考物之间的温度差、热流差,以表征所有与热效应有关的物理变化和化学变化,得到样品的相变信息。
[0037]
发明的有益效果
[0038]
与现有技术相比,本发明化合物通过简单重结晶,无需手性拆分,即可得到单一的磷(p)构型为r构型光学纯化合物,为固体性状,方便制备成口服制剂,有利于药物的口服吸收;本发明化合物在大鼠和beagle犬中灌胃给药的药代动力学数据显示,均可实现良好的口服吸收特征。
附图说明
[0039]
图1为化合物1的x射线单晶衍射图谱。
[0040]
图2为化合物2的x射线单晶衍射图谱。
[0041]
图3为化合物1的x射线粉末衍射图谱。
[0042]
图4为化合物1的差示扫描量热法(dsc)图谱
[0043]
图5为化合物2的x射线粉末衍射图谱。
具体实施方式
[0044]
以下结合实施例用于进一步描述本发明,但这些实施例并非限制着本发明的范围。
[0045]
实施例
[0046]
实施例给出了式(i)所表示的代表性化合物的制备及相关结构鉴定数据。必须说明,下述实施例是用于说明本发明而不是对本发明的限制。
[0047]
化合物的结构是通过核磁共振(nmr)和/或质谱(ms)来确定的。
[0048]1h nmr图谱是用bruker仪器(400mhz)测定而得,化学位移用ppm表示。使用四甲基硅烷内标准(0.00ppm),测定溶剂为氘代二甲基亚砜(dmso-d6),氘代氯仿(cdcl3),氘代甲醇(cd3od),内标为四甲基硅烷(tms)。1h nmr的表示方法:s=单峰,d=双重峰,t=三重峰,m=多重峰,br=变宽的,dd=双重峰的双重峰,dt=三重峰的双重峰。若提供偶合常数时,其单位为hz。
[0049]
质谱是用lc/ms仪测定得到,离子化方式可为esi或apci。
[0050]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.15mm~0.2mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。
[0051]
柱层析一般使用烟台黄海硅胶200~300目硅胶为载体。
[0052]
hplc的测定使用安捷伦1260dad高压液相色谱仪(zorba x sb-c18 100x 4.6mm)。
[0053]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.15mm~0.20mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。
[0054]
在下列实施例中,除非另有指明,所有温度为摄氏温度。
[0055]
除非另有指明,各种起始原料和试剂来自市售或者是根据已知的方法合成,市售原料和试剂均不经进一步纯化直接使用,除非另有指明,市售厂家包括但不限于aldrich chemical company,abcr gmbh&co.kg,acros organics,韶远化学科技(上海)有限公司,国药集团药业股份有限公司,百灵威科技有限公司等公司等处购买。
[0056]
对化合物进行纯化,采用硅胶柱层析和薄层色谱法,其中洗脱剂体系选自:a:石油
醚和乙酸乙酯体系;b:二氯甲烷和甲醇体系;c:二氯甲烷:乙酸乙酯;d:石油醚:二氯甲烷体系;其中溶剂的体积比根据化合物的极性不同而不同,也可以加入少量的酸性或碱性试剂进行调节,如醋酸或三乙胺等。
[0057]
dsc图谱仪器型号:ta dsc-q20差示扫描量热仪;吹扫气:氮气;升速速率:10℃/min;温度范围:25℃-250℃。
[0058]
x-射线粉末衍射谱图仪器型号:bruker d8advance x-射线粉末衍射仪;射线:单色cu-kα-1射线(λ=1.54056);扫描方式:θ/2θ,扫描范围:3-40o;电压:40kv;电流:40ma。
[0059]
化合物的绝对构型是通过x-射线单晶衍射谱来确定的,使用的仪器型号:bruker d8venture x-射线衍射仪;射线:gakα(λ=1.34139);温度:170.0k。
[0060]
实施例1
[0061]
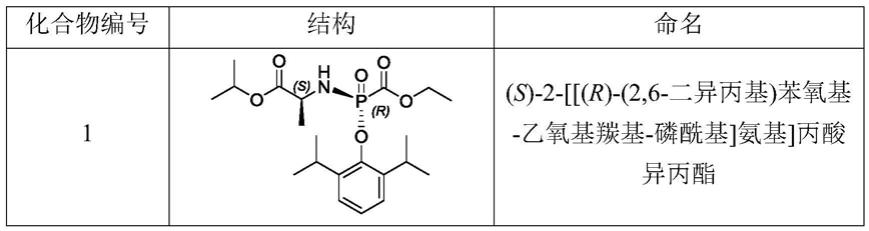
(s)-2-[[(r)-(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯(1)
[0062]
(s)-isopropyl 2-[[(r)-(2,6-diisopropylphenoxy)-(ethoxycarbonyl)phosphoryl]amino]propanoate(1)
[0063][0064][0065]
第一步
[0066]
乙氧羰基膦酸(1b)
[0067]
ethoxycarbonylphosphonic acid
[0068]
将二乙氧基磷酰甲酸乙酯1a(210.0g,1.0mol)溶于800ml乙腈中,滴入三甲基溴硅烷(460.0g,3.0mol),65℃反应2小时。反应结束后,减压浓缩,得到乙氧羰基膦酸1b,黄色液体(154.0g,1.0mol,产率100%)。
[0069]
第二步
[0070]
二氯磷酰甲酸乙酯(1c)
[0071]
ethyl dichlorophosphorylformate
[0072]
将甲氧羰基膦酸1b(154.0g,1.0mol)溶于650ml二氯甲烷中,氮气保护、0℃下依次滴加草酰氯(510.0g,4.0mol),dmf(10滴),升至室温反应2小时。反应结束后,减压浓缩,再加入二氯甲烷浓缩除去过量草酰氯(100ml x 3),所得液体减压蒸馏得到二氯磷酰甲酸甲酯1c,黄色液体(115.0g,0.61mol,产率61%)。
[0073]
第三步
[0074]
(s)-2-[[(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯(1e)
[0075]
isopropyl(s)-2-[[(2,6-diisopropylphenoxy)-(ethoxycarbonyl)phosphoryl]amino]propanoate
[0076]
将二氯磷酰甲酸甲酯1c(43.1g,0.227mol)溶于400ml二氯甲烷中,-20℃加入丙泊酚1d(40.0g,0.227mol)和三乙胺(46.0g,0.452mol)的二氯甲烷(200ml)溶液,保温反应30分钟,依次滴入l-丙氨酸异丙酯盐酸盐(38.0g,0.227mol)的二氯甲烷(200ml)溶液,三乙胺(46.0g,0.452mol),室温搅拌2小时。加水(500ml),饱和食盐水洗涤(200ml),无水硫酸钠干燥,减压浓缩,残留物用硅胶柱层析分离提纯洗脱剂体系a),得到(s)-2-[[(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯1e,无色油状物(43.0g,0.1mol,产率44%)。
[0077]
第四步
[0078]
(s)-2-[[(r)-(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯(1)
[0079]
(s)-isopropyl 2-[[(r)-(2,6-diisopropylphenoxy)-(ethoxycarbonyl)phosphoryl]amino]propanoate(1)
[0080]
将(s)-2-[[(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯1e(43.0g,100mmol)溶解于140ml正庚烷中,-20℃下放置7天,析出晶体,过滤,所得固体用少量冰正庚烷洗涤,烘干得到(s)-2-[[(r)-(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯1,白色固体(21.0g,49mmol,产率98%)。
[0081]1h nmr(400mhz,cdcl3):δ7.12-7.16(m,3h),5.01-5.04(m,1h),4.30-4.34(m,2h),4.13-4.17(m,1h),3.77-3.80(m,1h),3.37-3.44(m,2h),1.29-1.33(m,6h),1.21-1.28(m,18h)。
[0082]
31
p nmr(162mhz,cdcl3):δ-3.17
[0083]
lc-ms m/z=428[m+1]。
[0084]
化合物1的结晶样品的x-射线单晶衍射谱图见附图1,鉴定化合物1的绝对构型;化合物1的x-射线粉末衍射谱图见附图3,dsc谱图见附图4。
[0085]
上述母液旋干得到(s)-2-[[(s)-(2,6-二异丙基)苯氧基-乙氧基羰基-磷酰基]氨基]丙酸异丙酯1a,无色液体(21g,49mmol,产率98%)。
[0086]1h nmr(400mhz,cdcl3):δ7.11-7.16(m,3h),4.96-5.02(m,1h),4.28-4.31(m,1h),4.15-4.21(m,1h),3.81-3.87(m,1h),3.32-3.40(m,2h),1.40(d,j=7.2hz,3h),1.29(t,j=7.2hz),1.20-1.26(m,18h)。
[0087]
31
p nmr(162mhz,cdcl3):δ-3.79。
[0088]
lc-ms m/z=428[m+1]。
[0089]
实施例2
[0090]
(s)-2-[[(r)-(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯(2)
[0091]
(s)-isopropyl 2-[[(r)-(2,6-diisopropylphenoxy)-(methoxycarbonyl)phosphoryl]amino]propanoate(2)
[0092][0093][0094]
第一步
[0095]
二乙氧基磷酰甲酸甲酯(2b)
[0096]
methyl diethoxyphosphorylformate
[0097]
将亚磷酸三乙酯2a(50.0g,0.3mol)溶于250ml二氯甲烷中,0℃下滴入氯甲酸甲酯(28.4g,0.3mol),缓慢升至室温反应过夜。反应结束后,减压浓缩,得到二乙氧基磷酰甲酸甲酯2b,无色液体(59.0g,0.3mol,收率100%)。
[0098]
第二步
[0099]
甲氧羰基膦酸(2c)
[0100]
methoxycarbonylphosphonic acid
[0101]
将二乙氧基磷酰甲酸甲酯2b(59.0g,0.3mol)溶于250ml乙腈中,滴入三甲基溴硅烷(138.2g,0.9mol),65℃反应2小时。反应结束后,减压浓缩,得到甲氧羰基膦酸2c,黄色液体(42.1g,0.3mol,产率100%)。
[0102]
第三步
[0103]
二氯磷酰甲酸甲酯(2d)
[0104]
methyl dichlorophosphorylformate
[0105]
将甲氧羰基膦酸2c(42.2g,0.3mol)溶于200ml二氯甲烷中,氮气保护、0℃下依次滴加草酰氯(153.0g,1.2mol),dmf(10滴),升至室温反应2小时。反应结束后,减压浓缩,再
加入二氯甲烷浓缩除去过量草酰氯(50ml x 3),所得液体减压蒸馏得到二氯磷酰甲酸甲酯2d,黄色液体(20.0g,0.114mol,产率38%)。
[0106]
第四步
[0107]
(s)-2-[[(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯(2e)
[0108]
(2s)-isopropyl 2-[[(2,6-diisopropylphenoxy)-(methoxycarbonyl)phosphoryl]amino]propanoate(2e)
[0109]
将二氯磷酰甲酸甲酯2d(10.0g,56.5mmol)溶于100ml二氯甲烷中,-20℃加入丙泊酚1d(10.1g,56.5mmol)和三乙胺(11.5g,113.0mmol)的二氯甲烷(50ml)溶液,保温反应30分钟,依次滴入l-丙氨酸异丙酯盐酸盐(9.5g,56.5mmol)的二氯甲烷(50ml)溶液,三乙胺(11.5g,113.0mmol),室温搅拌2小时。加水(100ml),饱和食盐水洗涤(50ml),无水硫酸钠干燥,减压浓缩,残留物用硅胶柱层析分离提纯(洗脱剂体系a),得到(s)-2-[[(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯2e,无色油状物(6.0g,14.5mmol,产率25.7%)。
[0110]
第五步:
[0111]
(s)-2-[[(r)-(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯(2)
[0112]
(s)-isopropyl 2-[[(r)-(2,6-diisopropylphenoxy)-(methoxycarbonyl)phosphoryl]amino]propanoate(2)
[0113]
将(s)-2-[[(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯2e(6.0g,14.5mmol)2e溶解于20ml正庚烷中,-20℃下放置7天,析出晶体,过滤,所得固体用少量冰正庚烷洗涤,烘干得到(s)-2-[[(r)-(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯2,白色固体(2.9g,7.0mmol,产率97%)。
[0114]1h nmr(400mhz,cdcl3):δ7.12-7.16(m,3h),5.01-5.04(m,1h),4.12-4.18(m,1h),3.85(s,3h),3.74-3.77(m,1h),3.35-3.41(m,2h),1.32(d,j=6.8hz,3h),1.21-1.26(m,18h)。
[0115]
31
p nmr(162mhz,cdcl3):-3.40。
[0116]
lc-ms m/z=414[m+1]。
[0117]
化合物2的结晶样品的x-射线单晶衍射谱图见附图2,鉴定化合物2的绝对构型;化合物2的x-射线粉末衍射谱图见附图5。
[0118]
上述母液旋干得到(s)-2-[[(s)-(2,6-二异丙基)苯氧基-甲氧基羰基-磷酰基]氨基]丙酸异丙酯2a,无色液体(3.0g,7.2mmol,产率99%)。
[0119]1h nmr(400mhz,cdcl3):δ7.12-7.16(m,3h),4.98-5.01(m,1h),4.17-4.19(m,1h),3.86(s,3h),3.84-3.89(m,1h),3.30-3.37(m,2h),1.40(d,j=7.2hz,3h),1.20-1.26(m,18h)。
[0120]
31
p nmr(162mhz,cdcl3):δ-4.09。
[0121]
lc-ms m/z=414[m+1]。
[0122]
生物学评价
[0123]
测试例1、sd大鼠体内药代动力学研究
[0124]
试验动物:健康成年sd大鼠,雄性,220-270g,购于成都达硕实验动物有限公司。实验动物饲养于spf环境中,温度20-22℃,相对湿度51-55%,12小时/12小时明暗光照,自由
饮食饮水,适应性观察不少于3天后开始试验。
[0125]
药物配制:精密称取一定量受试化合物,加入dmso及solutol hs-15使溶解,再加入生理盐水,旋涡混合均匀。最终给药溶剂配比为dmso:solutol hs-15:生理盐水(10:10:80,v/v/v),所有受试化合物均在临用前新鲜配制。
[0126]
给药及检测:动物按体重随机分组,每组3只,给药前禁食不少于8小时,自由饮水,给药后4小时方可进食。各组大鼠分别静脉注射(iv)或灌胃(po)给予不同受试化合物溶液,于给药前及给药后不同时刻采集静脉血约0.1ml,肝素抗凝,12000转/分,4℃离心10分钟后收集血浆,于-80℃保存待测。采用lc-ms/ms法分别测定血浆中的原形药物(前药)及水解代谢物(原药)浓度,计算主要药动学参数,结果如表1所示:
[0127]
表1 sd大鼠药代动力学测试结果
[0128][0129]
备注:大鼠灌胃给予本发明化合物,各时刻血浆中均未检测到前药形式,仅检测丙泊酚并计算药动学参数。
[0130]
结论:化合物1和2大鼠灌胃给药后,体内主要以丙泊酚形式存在,根据丙泊酚血浆中暴露水平计算口服绝对生物利用度分别为23.9%和18.8%,表明具有良好的口服吸收特征。
[0131]
测试例2、beagle犬体内药代动力学研究
[0132]
试验动物:健康成年beagle犬3只,雄性,6-8kg,购于成都达硕实验动物有限公司。
[0133]
药物配制:精密称取一定量受试化合物,加入dmso及solutol hs-15使溶解,再加入生理盐水,旋涡混合均匀。最终给药溶剂配比为dmso:solutol hs-15:生理盐水(10:10:80,v/v/v),所有受试化合物均在临用前新鲜配制。
[0134]
给药及检测:动物给药前禁食不少于8小时,自由饮水,给药后4小时方可进食。试验分为3周期,第1、2、3周期依次灌胃(po)给予化合物1rp、2rp或静脉注射(iv)丙泊酚,于给药前及给药后不同时刻采集静脉血约1.0ml,肝素抗凝,12000转/分、4℃离心10分钟后收集血浆,于-80℃保存;清洗一周后进行下一周期给药。采用lc-ms/ms法分别测定血浆中的原形药物(前药)及水解代谢物(原药)浓度,计算主要药动学参数,结果如表2所示:
[0135]
表2 beagle犬药代动力学测试结果
[0136][0137]
备注:beagle犬灌胃给予本发明化合物,各时刻血浆中均未检测到前药形式,仅检测丙泊酚并计算药动学参数。
[0138]
结论:化合物1和2犬灌胃给药后,体内主要以丙泊酚形式存在,根据丙泊酚血浆中暴露水平计算口服绝对生物利用度分别为87.1%和78.4%,表明具有良好的口服吸收特征。
[0139]
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1