L-谷氨酸端基多肽分子印迹聚合物磁性微球及其制备方法和应用与流程
本发明属于化学分析测试领域,尤其涉及l-谷氨酸端基多肽分子印迹聚合物磁性微球及其制备方法和应用。
背景技术:
酸、甜、苦和咸是公认的四种基本味觉,而鲜味则被认为是第五种特殊味觉。人体中鲜味受体存在于口腔组织和肠道中,在肠道营养和消化中起重要作用。传统研究认为鲜味是由l-谷氨酸单钠(msg)、琥珀酸二钠、核糖核苷酸肌苷-5-单磷酸(imp)和鸟苷-5-单磷酸盐(gmp)等物质引起。而进一步研究提出,一些小分子多肽也能引起类似鲜味,称为呈鲜多肽,如含谷氨酸的亲水性二肽或三肽。酿造酱油中存在一类分子量在500da以下的小分子多肽,这些小分子多肽能够显著提高食品整体鲜味强度,是酿造酱油中的风味物质。呈鲜多肽不仅存在于肉类、水果等天然食品中,还存在于一些人工加工的食品中,如酱油、发酵火腿等。呈鲜多肽因营养价值高、天然、安全等特点,已成为食品鲜味的研究热点。
近年来,关于多肽特异性富集的研究报道越来越多。基于特殊的多肽富集机制,如低丰度肽的疏水相互作用、磷酸肽的亲和相互作用(静电,配位和螯合作用)以及糖肽的亲水相互作用,科学家们设计出多种新型纳米材料。多种介孔、磁性、化学修饰的亲和纳米材料被开发并应用于磷酸化多肽的分离与富集。研究表明,金属亲和纳米材料具有丰富的活性亲和位点、可控形态、大比表面积和可修饰的表面特性,可实现对生物分子的选择性分离与吸附,特异性强,灵敏度高,其多功能分层结构有利于亲和材料与目标生物分子之间的相互作用,有效缩短生物分子转移距离(易于转移)并提供亲和力介导功能,因此成为生物富集领域热门材料。磁性尖晶石型铁氧体亲和材料具有优异的单分散性,金属离子亲和性和便利的可分离性等优点,已在生物富集领域获得了广泛应用。
固相微萃取由于集采样、萃取、浓缩及进样于一体,具有耗时少、操作简单、效率高、少溶剂等优点,已成为国内外样品前处理技术领域的研究热点,在环境、生物、食品及药物分析等领域中获得了广泛应用。分子印迹技术是一种制备对某一特定靶标分子具有专一性识别作用化合物的技术。分子印迹聚合物(molecularimprintingpolymers,mips)中的三维识别空腔对模板分子具有高选择性识别能力,赋予该聚合物特殊“记忆”功能,类似自然生物的识别系统。mips因具备稳定性好、制备简单、生物相容性好等特点,已成为固相微萃取涂层材料的研究热点。其中,表位印迹法可直接在经过修饰的载体材料表面进行聚合,通过控制印迹位点于材料表面从而增加印迹位点个数,提高印迹效率,因此越来越多地被应用于蛋白质和多肽等生物大分子的分离与富集。近年来,活性/可控自由基聚合法已成为高分子化学及其它相关学科和领域研究的热点。其优点在于可控制聚合物分子量,形成具有精确一级结构的聚合物(组成、尺寸分布、形状、序列分布及规整性、侧链及端基结构),聚合物的分子量可以按设计预测且分布窄。其中,可逆加成断裂链转移法(reversibleaddition–fragmentationchaintransfer,raft)因适用功能单体范围宽、反应条件温和、抗干扰能力高、可以实现多种方式聚合(如本体、溶液、乳液、悬浮等聚合方式)、合成的聚合物分子量分布窄且随时间成线型分布等优点,已被广泛应用于多种特定结构如块状、梳型、星型、嫁接型聚合物的合成。
但现有的技术中,用于检测氨基酸和多肽的分子印迹聚合物存在包覆不均匀、作用力单一、性能不稳定等缺点,且常采用分步制备法,需要对基底材料进行多次修饰后再依次连接模板分子、功能单体和交联剂,存在制备步骤繁琐、印迹位点少、印迹涂层厚度不均匀、洗脱困难等缺点,从而大大影响了检测准性和灵敏度。
技术实现要素:
基于此,本发明的目的在于,提供一种l-谷氨酸端基多肽分子印迹聚合物磁性微球及其制备方法和应用。以制备得到印迹涂层包覆均匀、厚度均一、可同时特异性吸附多种端基为l-谷氨酸的呈鲜多肽分子、以及基底材料与模板分子之间的亲和力更强的分子印迹聚合物。
为达到上述目的,本发明采用如下技术方案:
l-谷氨酸端基多肽分子印迹聚合物磁性微球的制备方法,包括以下步骤:s1:制备cufemno4磁性微球,于聚合溶剂中加入cufemno4微球、替代模板分子、raft试剂、功能单体、交联剂和引发剂进行反应,得到包覆有替代模板分子的分子印迹聚合物@cufemno4磁性微球;s2:磁性分离后,采用洗脱溶剂洗脱步骤s1所得分子印迹聚合物@cufemno4磁性微球,除去其中包埋的替代模板分子后,干燥得到分子印迹聚合物@cufemno4磁性微球。
相对于现有技术,利用本发明所述方法制备的分子印迹聚合物@cufemno4磁性亲和纳米微球,采用“一锅法”,操作简单、耗时少、效率更高。一方面,以cufemno4微球作为基底材料,不仅可利用自身金属离子与模板分子产生金属螯合作用,还可利用cufemno4微球的磁性,实现快速分离富集。另一方面,模板分子可与功能单体结合,形成磁性微球-模板分子-单体复合物,将模板分子固定于框架中。另外,通过加入raft试剂,可实现微球表面聚合物自主生长,得到厚度较薄且可控的mips膜,从而克服传统自由基聚合法制备的mips传质慢、平衡时间长、模板分子难以洗脱等问题,进一步提高萃取效率。
进一步地,所述替代模板分子为l-谷氨酸,所述功能单体为烯丙基硫脲,所述raft试剂为s,s'-二(α,α'-二甲基-α-乙)三硫代碳酸酯。l-谷氨酸分子不仅可以与金属离子产生金属螯合作用,且模板分子上的羧基和氨基可与单体烯丙基硫脲中硫脲基团产生氢键和分子间作用力,形成磁性微球-模板分子-单体复合物,将模板分子固定于框架中。
更进一步地,所述替代模板分子、功能单体和raft试剂用量摩尔比为(0.05~0.3):(0.05~0.3):(0.1~0.4)。
进一步地,所述交联剂为n,n'-亚甲基双丙烯酰胺;所述引发剂为四甲基乙二胺和硫酸铵。
进一步地,所述聚合溶剂为tris-hcl缓冲溶液;所述聚合溶剂体积为1~10ml,所述聚合溶剂ph为6.5~10.5。
进一步地,所述洗脱溶剂为2%tfa的乙腈水溶液,所述乙腈和水的体积比为(3~10):(1~5)。
进一步地,步骤s1中,将cufemno4微球、替代模板分子、raft试剂、功能单体、交联剂和引发剂分散于聚合溶剂中,超声分散后充氮气除氧,置于摇床中15~55℃下振荡聚合1~10h,得到包覆有替代模板分子的分子印迹聚合物@cufemno4磁性微球。
本发明所述的l-谷氨酸端基多肽分子印迹聚合物磁性微球的制备方法,cufemno4磁性亲和纳米微球不仅作为基底材料,还可利用自身cu2+和fe3+金属离子与替代模板分子中的氨基与羧基发生金属螯合作用从而实现对模板分子的吸附。本方法所采用的“一锅法”聚合,同时于聚合体系中加入模板分子、功能单体和raft试剂等物质,一方面,可使得体系中模板分子用量大大增加,有利于提高萃取量。另一方面,由于可逆加成断裂链转自由基聚合法可有效控制聚合反应程度,因此得到的mips@cufemno4微球表面分子印迹聚合物层的结合位点分布较为分散,部分结合位点裸露在印迹层上,更有利于模板分子的洗脱和再吸附,从而提高萃取容量和效率。
本发明还提供由上述制备方法制得的l-谷氨酸端基多肽分子印迹聚合物@cufemno4磁性微球,所述分子印迹聚合物@cufemno4微球具有l-谷氨酸印迹聚合物刚性识别空穴。模板分子上的羧基和氨基可与cufemno4微球中的金属离子产生金属螯合亲和作用,同时与单体烯丙基硫脲中硫脲基团形成氢键和分子间作用力,形成磁性微球-模板分子-功能单体复合物。raft聚合后实现微球表面聚合物自主生长,得到厚度适宜且可控的mips膜。
本发明还提出按照上述制备方法所得的l-谷氨酸端基多肽分子印迹聚合物@cufemno4磁性微球在分离和检测酱油中呈鲜多肽的应用。
为了更好地理解和实施,下面结合附图详细说明本发明。
附图说明
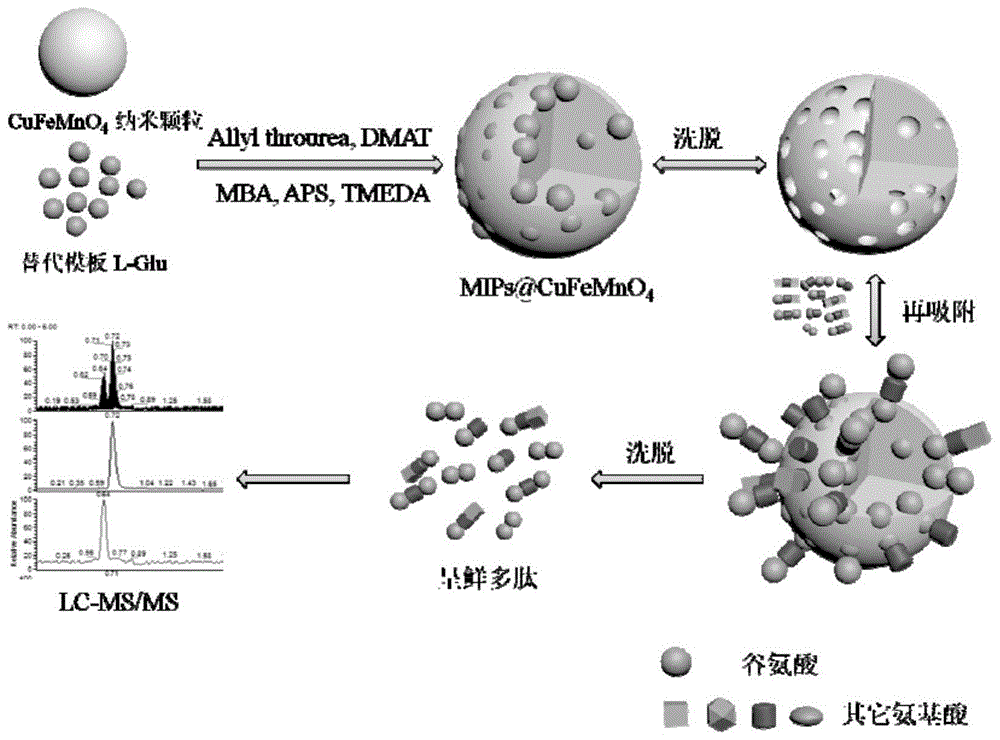
图1是本发明l-谷氨酸端基多肽分子印迹聚合物磁性微球制备过程以及应用过程示意图。
图2是本发明cufemno4微球(a)和mips@cufemno4微球(b)的透射电镜图。
图3是本发明cufemno4微球和mips@cufemno4微球的红外光谱图。
图4是本发明cufemno4微球和mips@cufemno4微球的x射线衍射图。
图5是本发明cufemno4(a)、nips@cufemno4微球(b)和mips@cufemno4微球(c)的热重分析图。(其中,nips@cufemno4微球除在制备过程中不添加模板分子以外,其余步骤与mips@cufemno4微球的制备完全相同。)
图6是本发明cufemno4微球和mips@cufemno4微球的分别对不同呈鲜多肽的选择性萃取容量图。
图7是本发明的mips@cufemno4微球和现有技术中的mips@sio2微球对端基为谷氨酸的二肽和三肽的萃取容量图。
图8是采用本发明制备的l-谷氨酸端基多肽分子印迹聚合物@cufemno4磁性亲和纳米微球对酱油进行预处理,和不采用上述微球对酱油进行预处理的液相色谱图。
具体实施方式
下面对本发明作进一步详细描述。
本发明提供一种l-谷氨酸端基多肽分子印迹聚合物磁性微球及其制备方法和应用。所述制备方法包括如下步骤:
s1:制备cufemno4磁性微球,于聚合溶剂中加入cufemno4微球、替代模板分子、raft试剂、功能单体、交联剂和引发剂进行聚合反应,得到包覆有替代模板分子的分子印迹聚合物@cufemno4磁性微球。
s2:磁性分离后,采用洗脱溶剂洗脱步骤s1所得分子印迹聚合物@cufemno4磁性微球,除去其中包埋的替代模板分子后,干燥得到分子印迹聚合物@cufemno4磁性微球。
其中,所述替代模板分子为l-谷氨酸,所述功能单体为烯丙基硫脲,所述raft试剂为s,s'-二(α,α'-二甲基-α-乙)三硫代碳酸酯。所述替代模板分子、功能单体和raft试剂用量摩尔比为(0.05~0.3):(0.05~0.3):(0.1~0.4)。
所述cufemno4磁性微球采用水热法合成,所述cufemno4为一种多功能磁性纳米亲和材料,利用亲和材料中的cu2+和fe3+与多肽中羧基和胺基的强配位作用,以其为载体,采用“一步法”在cufemno4纳米材料表面进行分子印迹。
优选的,本发明所述交联剂为n,n'-亚甲基双丙烯酰胺;所述引发剂为四甲基乙二胺和硫酸铵。所述洗脱溶剂为2%tfa的乙腈水溶液,所述乙腈和水的体积比为(3~10):(1~5)。
以下详述本发明的一种l-谷氨酸端基多肽分子印迹聚合物@cufemno4磁性亲和纳米微球的制备方法。请参阅图1,图1展示了本发明中l-谷氨酸端基多肽分子印迹聚合物@cufemno4磁性亲和纳米微球的制备过程示意图。所述制备方法包括以下步骤:
s1分子印迹聚合物@cufemno4磁性亲和纳米微球
准确称取5mgcufemno4微球于20ml圆底烧瓶中,加入7.35~44.1mgl-谷氨酸、5.8~34.8mgs,s'-二(α,α'-二甲基-α-乙)三硫代碳酸酯和2.82~5.68mg烯丙基硫脲,加入12mgn,n'-亚甲基双丙烯酰胺、17μl浓度为0.2499mg/ml的过硫酸铵和1μl四甲基乙二胺溶液分散于ph为6.5~10.5tris-hcl缓冲溶液中,所述tris-hcl缓冲溶液体积为1~10ml。超声分散后充氮气除氧,置于摇床中15~55℃下振荡聚合1~10h,得到包覆有替代模板分子的mips@cufemno4微球。
s2洗脱模板分子
磁分离后,用2%tfa的乙腈水溶液对聚合微球进行洗脱,洗脱液中乙腈和水的体积比为(3~10):(1~5)(ml/ml)。洗脱除去包埋于聚合物中的替代模板分子,干燥后得到具有l-谷氨酸印迹空穴的分子印迹聚合物@cufemno4磁性纳米微球。
通过上述制备方法所制得的l-谷氨酸端基多肽mips@cufemno4微球,具有l-谷氨酸印迹聚合物刚性识别空穴。下面是对l-谷氨酸端基多肽mips@cufemno4磁性亲和纳米微球进行的分析与应用。
为方便对比说明,除在制备过程中不添加替代模板分子l-谷氨酸以外,采用同样的方法制备得到非印迹聚合物微球(nip@cufemno4),用于与本实施例中制得的l-谷氨酸端基多肽分子印迹聚合物@cufemno4磁性亲和纳米微球(mip@cufemno4)对比分析。
请参阅图2,其是是本发明cufemno4、mips@cufemno4微球的透射电镜图。从图中可以看出,cufemno4颗粒近似球形,大小分布较为均匀,粒径约为7~12nm。在印迹聚合后,mips@cufemno4微球变大,粒径约为10~17nm。说明已成功在微球表面接上印迹聚合膜层。且聚合后mips@cufemno4微球仍能保持较好的分散性。
请参阅图3,其是本发明的cufemno4和mips@cufemno4微球的红外光谱图。通过对比可以发现,经过印迹后,化合物组成发生了明显变化。cufemno4属于尖晶石结构,其振动频率一般取决于八面体中的阳离子和氧之间的键作用力,与质量无关,频率较高的跃迁主要与八面体振动有关。根据这一判断,初步推断3425cm-1附近的吸收是八面体中fe-o振动所致,572cm-1附近红外吸收主要由mn2+在晶体中的不对称伸缩振动引起。印迹聚合物中3060cm-1属于羧基o-h键伸缩振动,1640cm-1为羧酸中羰基c=o伸缩振动;2950cm-1为c-h伸缩振动;1520cm-1为仲酰胺c=o的特征峰和硫脲中-n-c=s的伸缩振动峰;1225cm-1和703cm-1为硫代酯中c=s和c-s的伸缩振动峰。研究结果表明已在cufemno4纳米粒子表面成功聚合上一层印迹聚合物。
请参阅图4,其是本发明cufemno4和mips@cufemno4微球的x射线衍射图。制备的cufemno4中出现了8个衍射峰,分别为2θ=18.1,29.9,35.3,43.1,53.1,56.6,62.2和74.0°,对应了不同的晶面(111),(220),(311),(400),(422),(511),(440)和(533)。其衍射峰都与cufemno4的衍射峰(标准卡片jcpdsno.20-0358.27)相吻合,表明已成功合成尖晶石结构的cufemno4纳米颗粒,晶形较好。而图中蓝色标记的衍射峰2θ=43.1,50.4和74.0°归因于立方结构cu的衍射(111),(200)和(220)(标准卡片jcpdsno.04-0836)。11fe3++16cu2++mn2++3h6c2o2+10h2o→cufemno4+6cu+9cu++10fe2++6co2+38h+,乙二醇将部分cu2+还原成cu,cu相进入cufemno4晶格,从而形成整个晶体结构。这也就解释了在cufemno4晶体中存在cu的原因。经过印迹后,mips@cufemno4微球中cu和cufemno4特征强度减弱,但晶型结构没有发生改变,说明表明已成功印迹上一层分子印迹聚合物层。
请参阅图5,其是本发明cufemno4(a)、nips@cufemno4(b)和mips@cufemno4(c)微球的热重分析图,总质量损失分别为8.72%、17.89%和24.08%。通过对比发现其失重主要分为2个阶段,100~450℃和450~800℃。100~450℃阶段,cufemno4、nips@cufemno4和mips@cufemno4微球的失重率分别为7.10%、12.44%、15.14%。相对于cufemno4微球,聚合后nips@cufemno4和mips@cufemno4微球表现出更大的质量损失,其归因于微球表面聚合物壳层的质量丢失。其中,mips微球失重明显比mips微球失重多,是因为已经吸附在核心颗粒表面上的模板分子可促进表面接枝聚合,因此印迹膜层更厚。在450~800℃之间,nips和mips仍有约5.45%和8.49%的质量丢失。印迹聚合过程中由于金属与l-谷氨酸和raft试剂之间存在金属螯合作用,会有部分物质进入cufemno4晶格并在晶格中聚合,对聚合物起到了一定的保护作用,因此表现为高温失重。因此,通过热重曲线可进一步证明分子印迹层已成功在微球表面聚合。
请参阅图6,其是本发明nips@cufemno4和mips@cufemno4微球对不同呈鲜多肽的选择性萃取容量图。其中,选取的多肽依次为天门冬氨酸-甘氨酸(asp-gly)、谷氨酸-甘氨酸(glu-gly)、谷氨酸-谷氨酸(glu-glu)、丝氨酸-甘氨酸-丝氨酸(ser-gly-ser)、谷氨酸-甘氨酸-丝氨酸(glu-gly-ser)、谷氨酸-甘氨酸-谷氨酸(glu-gly-glu)。浓度均为0.01mg/ml。对于二肽asp-gly、glu-gly和glu-glu,mips@cufemno4微球的萃取量分别为0.25、0.12、0.43μmol;nips@cufemno4微球萃取量分别为0.027、0.073和0.16μmol。其中,两端都带谷氨酸的二肽glu-glu萃取量最高,是其它端基带一个或没有带谷氨酸二肽的1.73~3.56倍。对于三肽glu-gly-glu、glu-gly-ser和ser-gly-ser,mips@cufemno4微球萃取量分别为0.017、0.025和0μmol。mips@cufemno4微球对二肽的萃取量远高于三肽,说明由于印迹位阻的影响,二肽会更易于进入印迹空穴。mips@cufemno4微球对端基带一个或一个以上谷氨酸的呈鲜多肽的印迹因子为1.33~4.63,说明采用本发明的方法制得mips@cufemno4微球对端基含谷氨酸的呈鲜多肽具有较好的选择性。
请参阅图7,其是本发明制备的mips@cufemno4微球与和现有技术中mips@sio2微球对端基含谷氨酸的二肽或三肽的萃取印迹因子对比图;从图中可以看出,相同萃取条件下,mips@cufemno4微球对含谷氨酸的二肽或三肽的萃取选择性由优于mips@sio2微球。采用常规的sio2微球为基底材料,其表面积有限,且需要经过分步修饰,步骤繁琐,大大限制了目标物的萃取率。而本发明以cufemno4磁性亲和纳米微球为基底材料,不仅可利用自身金属离子与模板分子产生金属螯合作用,还可利用cufemno4微球的磁性,实现快速分离富集,提高萃取效率。采用“一锅法”聚合,模板分子和单体是同时加入反应体系中的,体系中模板分子的用量大大增加,得到的mips@cufemno4微球表面分子印迹聚合物层的结合位点分布较为分散,部分结合位点裸露在印迹层上,更有利于模板分子的洗脱和再吸附,从而萃取容量和效率更高。
请参阅图8,其是采用本发明制备的端基为l-谷氨酸的分子印迹聚合物@cufemno4磁性亲和纳米微球对酱油进行预处理和不采用上述微球对酱油进行预处理以及不采用上述微球对标准多肽参照溶液(非酱油)进行预处理的液相色谱图。经对比发现,未经mips@cufemno4微球萃取的酱油实际样色谱图基线较为复杂,杂峰多,严重影响目标物的检测。而经过mips@cufemno4微球萃取的酱油实际样色谱图可明显观察到杂质峰变少,检测到端基为谷氨酸的两种呈鲜多肽glu-gly和glu-glu,说明mips@cufemno4微球萃取可对样品实现富集作用。
相对于现有技术,利用本发明所述方法制备的分子印迹聚合物@cufemno4磁性亲和纳米微球具有表面包覆均匀、印迹聚合物分子量分布较窄、萃取性能稳定、萃取选择性高等优点。由于多种呈鲜多肽的端基为l-谷氨酸,因此可实现在水环境下对多种呈鲜多肽的选择性吸附,降低了工作量。另外,传统方法制备的mips@sio2微球模板分子与单体间作用力单一。而本方法制备的印迹微球中除了功能单体与替代模板分子之间的分子间作用力,模板分子上的羧基和氨基还可与微球中的金属离子产生金属螯合亲和作用,替代模板分子结合力显著增强,能够更加稳定地结合于聚合物的表面,印迹位点有效增多,以尽量多地甚至是全部结合酿造酱油中的端基为谷氨酸的呈鲜多肽,提高检测的准确性。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!