基于Taqman低密度芯片的胶质瘤EM/PM分子分型方法及其应用与流程
本发明涉及一种基于taqman低密度芯片(taqmanlowdensityarray)的胶质瘤em/pm分子分型方法及其应用。
背景技术:
弥散性胶质瘤(diffuseglioma)是成人中枢神经系统中最好发,也是最为致命的恶性肿瘤。尽管经过了数十年的不懈努力,胶质瘤的治疗至今仍然未能取得实质性突破。最常见的一类高恶性度胶质瘤——多形性胶质母细胞瘤(glioblastomamultiform,gbm)患者即使经手术切除肿瘤并同步接受目前最先进、最积极的放化疗,大多依然在一年内迅速复发进展、死亡。
无论在临床上及生物学上,胶质瘤皆具有极高的异质性。对胶质瘤进行合理的分型是改善其治疗效果的基础。传统的胶质瘤分型以组织学特征为参考,根据胶质瘤细胞与正常神经系统中各类型胶质细胞间假定的相似性,将成人弥散性胶质瘤分为星形细胞瘤、少突胶质细胞瘤以及混合型的少突星形细胞瘤,并依据核分裂、核密度、坏死程度以及血管增生等恶性指标进一步将胶质瘤分为ii-iv级。其中最恶性的为iv级星形细胞瘤通常,又被称为胶质母细胞瘤,约占成人胶质瘤的50%。形态学的诊断具有主观局限性,且不紧密结合胶质瘤的发病机制,阻碍了胶质瘤诊疗水平的提升。为克服这一缺陷,2016版的who胶质瘤分型指南将分子遗传学指标idh突变及1p/19q联合缺失等纳入分型指标,用以辅助组织病理学诊断。这两项指标的加入提高了胶质瘤诊断的客观性,以及风险分层的准确性。具有这两项变异的患者具有更好的预后,而且能够从放疗联合pcv(p:甲基苄肼,c:洛莫司汀,v:长春新碱)化疗的治疗方案中获益。
idh突变及1p19q联合缺失的肿瘤可能代表与少突胶质细胞祖细胞(opc)具有相似表达谱的一类胶质瘤的起始性基因组变异。然而胶质瘤的基因组变异并非稳定不变,而是处于动态进化中。在这一过程中,胶质瘤会不断积累越来越多的拷贝数变异(cnv,copynumbervariation)及单核苷酸突变(snv,singlenucleotideviraition)。这些变异与胶质瘤的分子病理及患者临床表现具有密切关联。事实上,一部分idh突变的患者因为细胞周期基因的激活而预后较差。此外,与肿瘤细胞学起源相关的表达谱特征也是决定肿瘤表型以及患者预后的重要因素之一。最近的一项研究发现,部分idh野生型、具有毛细胞星形细胞瘤(pilocyticastrocytoma,pa)基因表达特征的成人胶质瘤患者的预后远好于其它idh野生型患者。综上,以idh突变及1p19q联合缺失在预后评估及指导治疗方案选择方面尚且不足,建立一个独立于形态学,能捕捉胶质瘤与发育过程中的正常神经系统细胞间的转录组相似性的分型方案,有助于风险分层准确性的进一步提升以及更具针对性的治疗方案设计。
肿瘤的细胞学起源对其临床表型及治疗应答具有决定性的作用。基因表达谱(gep)蕴藏着肿瘤的细胞学起源信息,基于基因表达谱的分子分型有望将胶质瘤分型为具有独特细胞学起源的分子亚型,进而指导胶质瘤的风险评估和治疗方案选择。目前胶质瘤领域内已有数个gep分型。但是,它们普遍采取无偏差、无假说的分型方案,纯粹通过统计学方法筛选分类基因。分类基因与分子病理之间的联系、分子亚型与神经发育谱系之间的相似性都未能清楚地阐明,其指导的分子分型研究始终停滞于数据库分析层面,尚未有转化为个体化诊断工具的报道。
技术实现要素:
本发明的目的是提供一种基于taqman低密度芯片的胶质瘤em/pm分子分型方法及其应用。
第一方面,本发明要求保护检测特定基因表达量的物质和存储有em/pm分型预测模型的介质在制备产品中的应用;所述产品的功能为胶质瘤分型或预测胶质瘤患者生存期;
所述特定基因为44个基因;
所述44个基因分为em基因群、pm基因群和emlowpmlow基因群;
所述em基因群由如下12个基因组成:acss3、cdkn2c、egfr、elovl2、hs3st3b1、itgb8、nes、sec61g、socs2、uhrf1、vav3和pdgfa;
所述pm基因群由如下28个基因组成:c1ql1、chd7、csnk1e、dll3、etv1、klrc3、lphn3、lppr1、marcks、mmp16、myt1、nav1、nlgn1、nova1、nxph1、olig1、olig2、patz1、pcgf2、pdgfra、rfx7、sox6、sox8、tacc2、tshz1、zeb1、znf22和znf462;
所述emlowpmlow基因群由如下4个基因组成:enpp2、gabra1、mal和mog;(上述四个基因高表达于正常脑组织,是非肿瘤脑组织的标记基因)
所述em/pm分型预测模型按照包括如下步骤的方法获得:
(1)分别检测n个样本的特定基因的表达量;
(2)取步骤(1)获得的所有样本的特定基因表达量的数据,通过一致性聚类算法将每个胶质瘤样本分为em型、pm型或emlowpmlow型;
(3)取步骤(1)获得的所有样本的特定基因表达量的数据和步骤(2)获得的所有样本的分型结果,通过支持向量机算法构建以特定基因表达量为输入以分型归属概率为输出的模型,即为em/pm分型预测模型;
所述分型归属概率即某一样本为em型的概率、为pm型的概率和为emlowpmlow型的概率。
所述n个样本为有统计学意义的数量的样本。所述n个样本可为胶质瘤样本。如果样本中emlowpmlow型数量过少导致无法准确分型,可添加非肿瘤样本进行分析。
进一步地,本发明要求保护检测特定基因表达量的物质、存储有em/pm分型预测模型的介质和记载有特定方法的载体在制备产品中的应用;所述产品的功能为胶质瘤分型或预测胶质瘤患者生存期;所述特定方法为方法a或方法b;
所述特定基因为前文所述的特定基因;
所述em/pm分型预测模型按照前文所述的方法获得;
所述方法a为获得待测患者胶质瘤em/pm分型的方法,包括如下步骤:
(a1)检测待测胶质瘤患者特定基因的表达量;
(a2)将待测胶质瘤患者特定基因的表达量输入所述em/pm分型预测模型,由模型输出待测胶质瘤患者的分型归属概率;
(a3)根据待测胶质瘤患者的分型归属概率,获得待测胶质瘤患者的分型结果;判断标准为:若待测胶质瘤患者属于某一类型的概率大于70%,则判断患者属于该类型,若待测患者属于任一类型的概率均低于70%,则该患者为难以确定类型的非典型患者;
所述方法b为预测胶质瘤患者生存期的方法,包括如下步骤:
(a1)检测待测胶质瘤患者特定基因的表达量;
(a2)将待测胶质瘤患者特定基因的表达量输入所述em/pm分型预测模型,由模型输出待测胶质瘤患者的分型归属概率;
(a3)根据待测胶质瘤患者的分型归属概率,获得待测胶质瘤患者的分型结果;判断标准为:若待测胶质瘤患者属于某一类型的概率大于70%,则判断患者属于该类型,若待测患者属于任一类型的概率均低于70%,则该患者为难以确定类型的非典型患者;
(a4)根据待测胶质瘤患者的分型结果按照如下标准对其进行生存期预后:em型患者的生存期低于pm型患者的生存期。
第二方面,本发明保护用于胶质瘤分型或预测胶质瘤患者生存期的试剂盒,包括检测特定基因表达量的物质和存储有em/pm分型预测模型的介质;
所述特定基因为前文所述的特定基因;
所述em/pm分型预测模型按照前文所述的方法获得。
所述试剂盒中还可以包括记载前文所述的方法a或方法b的载体。
在上述第一方面和第二方面中,所述检测特定基因表达量的物质为taqman低密度芯片;所述taqman低密度芯片上固定有用于扩增所述44个基因的引物对。所述taqman低密度芯片上还固定有用于扩增gapdh、actb、ubc和18srrna的引物对;所述gapdh、actb和ubc为内参基因;所述18srrna为pcr体系质控基因。
在第二方面中,当所述检测特定基因表达量的物质为taqman低密度芯片时,所述试剂盒还包括使用taqman低密度芯片检测待特定基因表达量时所需的其他试剂。
第三方面,本发明要求保护一种用于制备em/pm分型预测模型的系统组合,包括系统甲、系统乙和系统丙;由所述系统甲检测n个样本特定基因的表达量,输出每个样本特定基因的表达量的数据;由系统乙接收系统甲输出的所有样本的特定基因表达量的数据,通过一致性聚类算法将每个样本分为em型、pm型或emlowpmlow型;由系统丙接收取系统甲输出的所有样本的基因表达量的数据和系统乙输出的所有样本的分型结果,通过支持向量机算法构建以特定基因表达量为输入以分型归属概率为输出的模型,即为em/pm分型预测模型;所述分型归属概率概率即某一样本为em型的概率、为pm型的概率和为emlowpmlow型的概率。所述特定基因为前文所述的特定基因。所述n个样本为有统计学意义的数量的样本。所述n个样本可为胶质瘤样本。如果样本中emlowpmlow型数量过少导致无法准确分型,可添加非肿瘤样本进行分析。
第四方面,本发明要求保护一种用于制备em/pm分型预测模型的方法,包括如下步骤:
(1)分别检测n个样本特定基因的表达量;所述特定基因为前文所述的特定基因;
(2)取步骤(1)获得的所有样本的特定基因表达量的数据,通过一致性聚类算法将每个胶质瘤样本分为em型、pm型或emlowpmlow型;
(3)取步骤(1)获得的样本的特定基因表达量的数据和步骤(2)获得的所有样本的分型结果,通过支持向量机算法构建以特定基因表达量为输入以分型归属概率为输出的模型,即为em/pm分型预测模型;
所述分型归属概率即某一胶质瘤样本为em型的概率、为pm型的概率和为emlowpmlow型的概率。
所述n个样本为有统计学意义的数量的样本。所述n个样本可为胶质瘤样本。如果样本中emlowpmlow型数量过少导致无法准确分型,可添加非肿瘤样本进行分析。
以上各方面中,所述em/pm分型预测模型的建立使用python平台的机器学习包scikit-learn完成,使用python平台的机器学习包scikit-learn构建所述模型的具体方法如下:
导入numpy、pandas、scipy、matplotlib、itertools以及sklearn等分析所需的python模块;
从csv、txt等格式的文件中读取训练样本的基因表达谱;
将表达量数值做log2转化;
将表达数值进行归一化处理;
从csv、txt等格式的文件中读取训练样本的分型标签,将其二分化处理;
向训练集中添加随机化的噪音数据,避免过拟合;
将样本随机划分为10份;
使用支持向量机(svm)算法构建分类模型;
并建立10折交叉验证模型;
利用模型进行100次交叉验证,计算平均的准确率、精准度及召回率;
跟据交叉验证的结果,使用网格搜索寻找最佳svm超参数,或得使准确率等尽量高的超参数;
从csv、txt等格式的文件中读入验证集样本数据;
将表达量数值做log2转化;
将表达数值进行归一化处理;
从csv、txt等格式的文件中读取训练样本的分型标签,将其二分化处理;
使用在训练集中构建的模型预测验证集中样本的亚型;
比较验证集样本的预测亚型与已有分型标签,计算假阳性(fpr)、真阳性率(tpr);
根据每一亚型的fpr、tpr计算每一亚型的roc及auc;
计算验证集中总体的宏平均roc及auc;
绘制roc图;
观察roc图发现模型在验证集中具有很高的准确率,因此可应用于新样本的分型;
导入joblib模块;
使用joblib模块将对应与分类模型的对象写入到本地em_pm_svm.model模型中。
以上各方面中,所述特定基因的表达量为相对表达量。
所述相对表达量为基于内参基因的相对表达量。计算特定基因与3个内参基因的几何平均值之间的差值(δct),以2-δct作为特定基因的相对表达量。
所述内参基因为gapdh、actb和ubc。
第五方面,本发明保护下述任一产品;
(a)存储有前文所述em/pm分型预测模型的介质;
(b)前文所述的taqman低密度芯片。
进一步地,本发明还保护所述产品在制备用于患者胶质瘤分型和/或预测胶质瘤患者生存期的产品中的应用。
本发明提供了一种可具体实施em/pm分子分型的taqman低密度芯片(taqmanlowdensityarray,tlda)产品及配套试剂盒,以及基于支持向量机(supportvectormachine,svm)的em/pm分型算法构建的模型。使用该芯片及模型可快速便捷地完成病人em/pm分型及个体化诊断,对患者给出风险分层及个体化用药的指导。相较于基于affymetrix芯片或mrnaseq的表达谱检测分型要7到10天才能完成,本发明的检测方法仅需在一个工作日内就能完成。同时,本发明采用的芯片及检测仪器均为实验室常备的检测仪器,不需再额外装备大型设备(例如affymetrix芯片检测系统或高通量测序仪器)。
附图说明
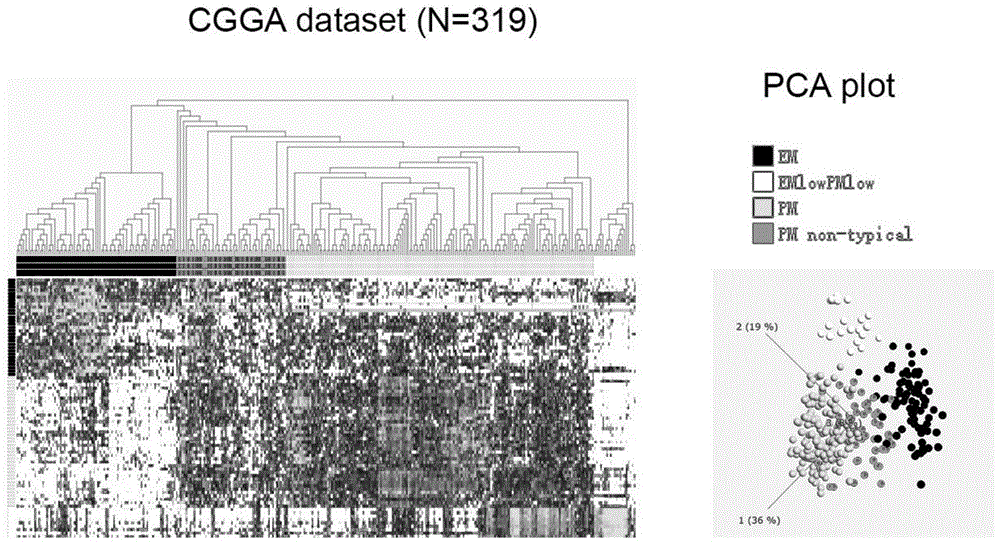
图1为cgga数据库中使用分型基因进行聚类分析的结果。
图2为使用非负矩阵分解(nmf)及一致性聚类算法对cgga数据库进行高稳定性分子分型以建立训练集样本分型标签的结果。
图3为选择cgga训练集中59例样本,使用tlda芯片检测表达谱,然后使用svm算法训练分类模型的结果。
图4为验证集中模型验证的效果。
图5为使用上述所建立的模型所预测的em及pm胶质瘤的生存曲线图。
具体实施方式
以下的实施例便于更好地理解本发明,但并不限定本发明。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。以下实施例中的定量试验,均设置三次重复实验,结果取平均值。
实施例1、训练样本库em/pm分子分型的建立
本发明的目标为实现单独患者的em/pm分子分型,欲实现此目的,需采取有监督(supervised)分类算法,而有监督分类算法则需要一组分型已知的样本作为训练样本集。中国脑胶质瘤基因组图谱(chinesegliomagenomeatlas,cgga)数据库中包含319例胶质瘤样本的mrna测序数据,部分上述样本还有肿瘤组织留存,可用于rna提取及tlda芯片检测,所得数据作为建立个体化em/pm分型预测模型的训练集。
首先通过对公共脑胶质瘤基因表达数据库rembradnt进行分析,计算候选em/pm分类基因的变异系数(cv),共筛选出了变异系数大的27个em基因、32个pm基因、9个标记emlowpmlow基因。此外,在rembrandt数据库中进行了全基因组共表达网络分析(wgcna),鉴定了15个在各em/pm分型中特异表达的基因网络中的枢纽基因。具体如表1所示。
基于所述319例样本的mrna表达数据,首先采取经典的层级聚类算法进行聚类分析并绘制热图。使用qlucore3.4(qlucoreab.)软件内置的层级聚类算法对来自cgga数据库中319样本进行层级聚类以及热图绘制。挑选出典型的样本,再进一步使用nmf算法降维,使用consensusclustring算法进行聚类,聚类算法为pam(partitioingaroundmedoid)以获得训练集样本稳定的亚型标签。具体实施由r软件的“nmf”包进行。进行聚类时使用的基因如表1所示。结果显示,利用所述层级聚类方法,根据所述基因的差异表达,可将这群样本清楚地分为三个主要的类型,分别是高表达em基因的em型、高表达pm基因的pm型以及高表达正常神经元或神经胶质细胞瘤基因的emlowpmlow型(图1)。层级聚类的结果可以进行大致的em/pm分子分型,但是层级聚类难以确定亚型的数目,而且层级聚类的结果不够稳定,不易重现。
一致性聚类(consensusclustering)是一种用于建立稳定的分型,并确定最佳的分型数目的算法。对上述319例样本,本发明的发明人采取了nmf算法重新进行降维分析,再联合使用一致性聚类对上述样本分型。结果如图2所示。图2a为分类数目从2到7时一致性聚类结果,可见当分型数目大于3时,分类的稳定性显著下降。图2b为三个评估最佳分型数目的参数,包括共表型系数(copheneticcoefficient)、离差(dispersion)、轮廓宽度(silhouettewidth,sw)。这些参数越高,表明分类效果越好,当这些参数下降时,就表明分型不再稳定。当k>3时,三个参数都骤然降低,表明k=3为比较理想的分类数目,可以将胶质瘤分为em、pm和emlowpmlow三个类型(图2b)。轮廓宽度(silhouettewidth,sw)也是评估样本是否为亚型内典型样本的一个参数。它表征样本与其属同一类型的所有其它样本的相似程度。sw取值[-1,1]之间。sw越低,样本的亚型典型性越低。当sw小于0时,提示其更类似其它类型中的样本。在后续的分析中,排除了sw值低于0.1的56例非典型样本,保留了263例样本。对这263例样本进行生存分析发现,em型的胶质瘤5年生存率为0,而pm和emlowpmlow型的胶质瘤在第五年时的生存率超过60%(图2c)。
实施例2、胶质瘤分型基因群的筛选和检测芯片的制备
一、胶质瘤分型基因群的筛选
使用表1所述基因设计了一个包含96基因的panel。
表1
将这些基因交由公司设计引物并制备成taqman低密度芯片。为初步评估该平台的实际表现,并基于实时荧光定量pcr数据进一步缩小候选基因的范围,挑选实施例1中所述263例样本中的40例典型的胶质瘤样本,使用该芯片进行了检测。结果发现在96个基因中,有6个基因(dmrta2、shox2、sox4、tnfrsf19、mobp、polr2f)的ct值很大或者是在大部分样本中都无扩增,这些基因被首先排除。
为进一步计算各基因的相对表达量,根据表达差异进一步筛选分类基因,首先对13个内参基因进行筛选。候选内参基因的评分由genorm算法给予每个候选内参基因的评分,评分越低,越适合作为内参基因。评分最低的3个被选为最终的内参基因,它们分别是actb、gapdh及ubc。以这三个基因的几何平均值为对照,计算剩下的77个分类基因的相对表达量,并比较它们在不同类型之间的差异,计算得到的p值及foldchange。根据foldchange、p值以及基因功能筛选出44个基因作为tlda平台的em/pm分型基因。这44个分类基因包含12个em基因,28个pm基因及4个emlowpmlow基因。12个em基因功能涉及到神经干细胞及早期星形胶质细胞分化、肿瘤迁徙及肿瘤微环境免疫调控;28个pm基因的功能涉及到少突胶质细胞祖细胞分化、脂代谢、染色质修饰和上皮-间质转化等。4个emlowpmlow基因中有两个标记正常成熟少突胶质细胞,另外两个分别标记正常星形胶质细胞及成熟神经元。这44个分型基因与gapdh、actb、ubc以及18srrna共同构成用于个体化em/pm分型的基因(表2)。
表2
二、taqman低密度芯片的制备
由lifetechnology公司设计用于扩增表3中每种基因的引物,由3m公司制备用于检测表2基因的taqman低密度芯片。
实施例3、利用tlda芯片进行胶质瘤的个体化分子分型
从实施例1中所述的263例样本中挑选了52例肿瘤样本,并加入7例对照正常脑组织样本作为训练样本集。随后使用实施例2中所述48基因taqman低密度芯片检测这些样本的基因表达谱。
检测方法如下:
1、提取样本的总rna,并反转录为cdna。
2、使用实施例2制备的taqman低密度芯片检测待测44个分型基因、3个内参基因及质控基因的表达量,所使用仪器为abi7900htfast实时荧光定量pcr系统。配制100μl的反应体系,内含:50μl2×taqmanuniversalpcrmastermixwithung(thermofisher,货号4352042)、48μl无rna酶水以及2μl模板cdna。然后将反应体系转移至低密度芯片的上样槽内,1500rpm快速离心一分钟,离心两次。使用封版机,通过封压金属膜,隔断反应槽,使进行扩增反应时样本间互不干扰。随后,将taqman低密度芯片放入实时荧光定量pcr仪中,按如下条件进行扩增:50℃ung孵育2分钟,随后进行40个循环的扩增,每一循环分为两步,分别是95℃变性15秒,60℃退火1分钟。40个循环结束后,再60℃孵育一分钟。反应过程中使用sds2.3软件采集数据。
3、将实时荧光定量pcr仪生成的sds格式的文件(abi7900htqpcr仪输出)导入到软件expressionsuitesoftwarev1.0.4中,每个基因的循环数(ct)阈值由软件自动判定。然后,计算待检测基因与3个内参基因的几何平均值之间的差值(δct),以2-δct作为分类基因的相对表达量。最后将各次实验采集到样本的em/pm分型基因的表达数据汇总在一起,进行归一化处理,形成用于后续步骤分析的基因表达数据。
对得到的基于tlda表达谱数据进行降维分析,用以直观观测这些基因的分型效果。主成分分析图表明,具有相同亚型的样本在空间上紧密的聚集在了一起,而具有不同类型的样本在空间上相互远离(图3a),初步表明了实时荧光定量pcr数据能够区分开各em/pm类型。但是要进行精确的个体化分型,需要应用有监督的聚类算法。在众多的有监督分类算法中,支持向量机算法在小样本中的效果较佳,不仅准确率高,而且不容易遭遇过拟合问题。
采取支持向量机算法,利用归一化之后的训练集数据来建立em/pm分型模型,数据的统计分析以及分类算法训练、验证以及样本分型的判别均使用python平台的机器学习包scikit-learn完成。使用c-svc中一对一并行比较算法在训练集进行多分类模型训练,并通过网格搜索方法优化参数,确定了支持向量机的最佳参数(cost=0.5,degree=3,高斯核函数,gamma=8),并得到基于svm的em/pm分型预测模型。该模型将计算样本属于每一类型的概率。若属于某一类型的概率大于70%,则判断样本属于该类型,若样本属于任一类型的概率均低于70%,则该样本为难以确定em/pm类型的非典型样本。
对于得到的模型,采用10折交叉验证计算其在训练集中的准确率、精确度及召回率三个评估模型效力的关键参数的值。100次交叉验证的计算结果发现,判断该分类模型效果优劣的三项指标的均值分别为83%,85%及82%(图3b)。上述结果表明,以tlda数据为训练数据,以svm为分类算法,可以得到较好的个体化分类模型。
为了确定建立的基于svm的em/pm分型预测模型是否适用于新的数据集,是否会在新样本、新平台上遇到过拟合或拟合不足的情况,另外建立一个包含35例样本的独立验证队列(含31例胶质瘤及4例非肿瘤脑组织)。
对于这35例样本,使用affymetrix的clariomd芯片检测其全转录组基因表达谱,由nmf算法确定31例样本中有9例em型胶质瘤、19例pm型胶质瘤及3例emlowpmlow型胶质瘤,四例非肿瘤脑组织也属于emlowpmlow型。主成分分析图(图4a)表明,依据来自全转录组芯片的数据及nmf无监督聚类算法的分型标签,具有相同亚型的样本在空间上紧密的聚集在了一起,而具有不同亚型的样本在空间上相互远离。
使用tlda芯片测量了这批样本44个em/pm分类基因的表达谱,使用em/pm分型预测模型对其进行了个体化分型,并将分型结果与全转录组芯片nmf算法所得分型进行对比。
结果表明,在35例样本中,有一例pm型胶质瘤被svm模型预测为em型胶质瘤,有一例emlowpmlow型胶质瘤被svm模型预测为pm型胶质瘤,其他33例样本的nmf分型与基于tlda数据的个体化分型均保持一致。roc(受试者工作曲线)分析显示em、pm及emlowpmlow型胶质瘤的auc(areaundercurve,曲线下面积)分别为0.86、0.85及0.91(图4b),auc是敏感度与特异性的综合反映,auc值越大,表明分型的灵敏度与特异性就越高。完美的分型auc值为1,当auc大于0.7时,就表明分型模型是有效的。
上述结果证明所训练的个体化em/pm分类模型在新的数据集中依然保持着较高的准确率,且独立于检测平台。
使用python平台的机器学习包scikit-learn构建所述模型的计算机语言如下:
利用训练集的模型预测亚型已知的在验证集本的亚型,检验预测模型的灵敏度与特异性
使用以下代码预测新样本的empm亚型
#首先读入待测样本的表达数据test.txt,并log2转化,然后归一化处理。
vdata=pd.read_csv('validation.csv')
validata=np.array(vdata[1:])
validata=np.log2(validata)
validata=preprocessing.scale(validata,axis=1)
y2=np.array(vdata[:1])
y2=y2[0].astype('int')
#然后使用先前构建的模型进行分型,y_score将返回样本属于每一亚型的概率y_score=classifier.fit(traindata,y2).decision_function(validata)
实施例4、实际样本检测
待测样本:122例em/pm类型未知的样本,获自北京天坛医院及瑞典哥德堡大学sahlgrenska医学院。
1、提取待测样本的总rna,并反转录为cdna。
2、使用实施例2制备的taqman低密度芯片检测待测44个分型基因、3个内参基因及质控基因的表达量,所使用仪器为abi7900htfast实时荧光定量pcr系统。配制100μl的反应体系,内含:50μl2×taqmanuniversalpcrmastermixwithung(thermofisher,货号4352042)、48μl无rna酶水以及2μl模板cdna。然后将反应体系转移至低密度芯片的上样槽内,1500rpm快速离心一分钟,离心两次。使用封板机,通过封压金属膜,隔断反应槽,使进行扩增反应时样本间互不干扰。随后,将taqman低密度芯片放入实时荧光定量pcr仪中,按如下条件进行扩增:50℃ung孵育2分钟,随后进行40个循环的扩增,每一循环分为两步,分别是95℃变性15秒,60℃退火1分钟。40个循环结束后,再60℃孵育一分钟。反应过程中使用sds2.3软件采集数据。
3、将实时荧光定量pcr仪生成的sds格式的文件(abi7900htqpcr仪输出)导入到软件expressionsuitesoftwarev1.0.4中,每个基因的循环数(ct)阈值由软件自动判定。然后,计算待检测基因与选出的多个内参基因的几何平均值之间的差值(δct),以2-δct作为分类基因的相对表达量。最后将各次实验采集到样本的em/pm分型基因的表达数据汇总在一起,进行归一化处理,形成用于后续步骤分析的基因表达数据。
4、将步骤3的数据输入实施例3建立的模型中,得到分型结果。分型结果为这122例样本中有39例em型胶质瘤,64例pm型胶质瘤以及18例emlowpmlow型胶质瘤。
5、按照个体化预测得到的em/pm分型标签进行主成分分析,结果表明,该模型所预测的同一类型样本明显地聚集到了一起。
对上述实施例4中的122个样本和实施例3中的35例样本(独立验证队列)的临床数据进行分析可以看到:em型胶质瘤患者的发病年龄高于pm型胶质瘤患者(em型胶质瘤患者59.1±12.4岁,pm型胶质瘤患者:46.1±13.5岁)。em型胶质瘤患者的生存时间显著低于pm型胶质瘤患者的。em型胶质瘤患者的一年、两年、三年生存率分别为55%,21%及17%,而pm型胶质瘤患者的一年、两年三年生存率分别为87%,71%,68%(图5)。
- 还没有人留言评论。精彩留言会获得点赞!