一种嘌呤衍生物的合成及应用的制作方法
本发明属于化合合成领域,特别涉及一种嘌呤衍生物的合成及应用。
背景技术:
甲基化修饰的通用甲基供体s-腺苷甲硫氨酸(sam)在多胺合成酶、n-酰基高丝氨酸内酯合成酶的作用下除生成多胺及n-酰基高丝氨酸内酯外均生成甲基硫腺苷(mta)。mta在大部分病原微生物等原核细胞内被甲基硫腺苷/s-腺苷高半胱氨酸核苷酶(sahn)裂解为腺嘌呤和5′-甲硫核糖(mtr),mtr随即被5′-甲硫核糖激酶磷酸化生成5′-甲硫核糖-1-磷酸(mtrp)。而在作为致病菌宿主的哺乳动物等真核生物细胞内,mta则是由专一性5′-甲硫腺苷磷酸化酶催化一步反应生成mtrp和腺嘌呤。随后mtrp经多步反应生成甲硫氨酸(met),met在sam合成酶的作用下又重新生成sam,从而完成整个amc甲基循环(activatedmethylcycle)。病原微生物和宿主细胞在amc甲基循环中mta节点的显著差异,使病原微生物催化mta裂解的sahn核苷酶成为抗感染药物的潜在靶点。
技术实现要素:
为弥补现有技术的不足,本发明提供了一种嘌呤衍生物的合成及应用。
本发明的发明构思是:由于mta不带生物素,所以mta不能与带有链和亲霉素的streptavidindonorbeads结合,因此需要在mta结构的基础上将生物素的结构引入到化合物中,而底物mta与sahn结合的中心位点是嘌呤碱基部分,于是发明人选择同样拥有嘌呤碱基的六氯嘌呤核苷进行结构的改造。发明人以六氯嘌呤核苷为起始物质,首先将9位上的氨基进行thp保护,再以性质稳定的
为实现上述目的,本发明采用以下技术方案:
一种嘌呤类化合物,具有如式ⅰ所示结构:
上述嘌呤类化合物的制备方法,包括如下步骤:
(1)首先将六氯嘌呤与对甲苯磺酸p-tsoh溶于四氢呋喃中,60℃油浴加热的情况下加入3,4-二氢-2h吡喃,在80℃条件下回流反应12h,反应结束后,利用旋转蒸发仪蒸干溶剂,萃取,干燥,柱层析分离提纯,得到化合物1;其中六氯嘌呤、对甲苯磺酸、3,4-二氢-2h吡喃的摩尔当量比为1:0.2:1.5。
(2)将化合物1溶于正丙醇后,加入乙二胺和n,n-二异丙基乙胺dipea油浴80℃回流反应3小时,利用旋转蒸发仪蒸干溶剂,过硅胶柱进行粗分离,得到的化合物2;其中化合物1与乙二胺、dipea的摩尔当量比为1:1.4:2。
(3)将生物素biotin用干燥好的二甲基甲酰胺dmf溶解后,加入n-羟基硫代琥珀酰亚胺nhs和edci盐酸盐在室温下反应7h后,向上述反应混合物中加入dipea和化合物2并继续在常温下反应3h,冷冻干燥去除溶剂后,用柱层析分离纯化的化合物3;其中,化合物2、生物素、n-羟基硫代琥珀酰亚胺、edci盐酸盐、n,n-二异丙基乙胺的摩尔当量比为1:1.2:1.2:3:5。
(4)将化合物3与吡啶对甲苯磺酸盐ppts溶解于dmf中,常温反应15h。冷冻干燥溶剂,用柱层析分离纯化得到化合物4。化合物3与吡啶对甲苯磺酸盐的摩尔当量比为1:0.2。
本发明所述的edci盐酸盐为碳化二亚胺edci盐酸盐,即1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐)。
本发明同时请求保护上述化合物在筛选抑制剂制备上的应用。
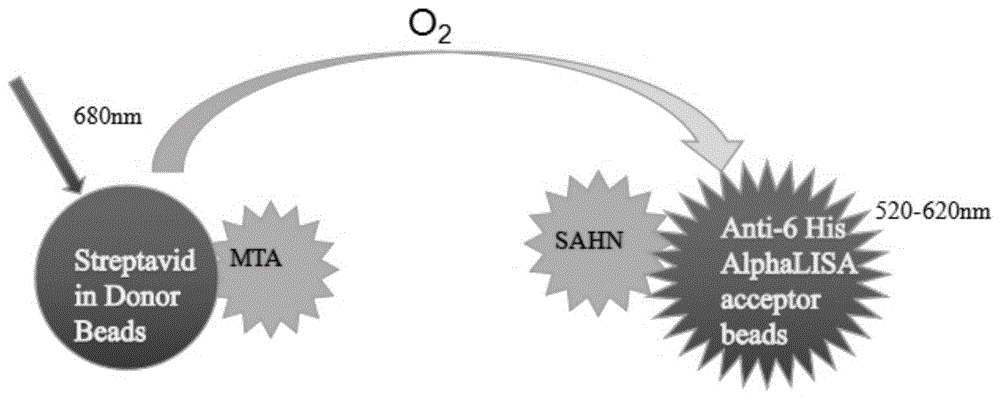
本发明将生物素的结构引入到嘌呤结构中,得到的化合物嘌呤衍生物和5′-甲基硫腺苷的结构类似并且带有生物素,可以替代mta,构建出一个基于alphalisatechnology筛选抑制剂的体系,有选择性的杀死入侵的微生物。为今后合成带有生物素结构的嘌呤类衍生物提供了新的合成方法,也为开发具有更高特异性和稳定性的抑制剂,以及免疫学研究提供了新型化合物。
有益效果:
(1)本发明具有反应温度低,反应时间短,纯化方法简单,产率高等优点。
(2)本发明设计的嘌呤类化合物结构以酰胺键替代氨基键,分子化学稳定性更高。
(3)本发明合成的化合物成本低,具有一定的经济效益。
附图说明
图1alphalisatechnology的筛选体系;
图2为本发明化合物1的氢谱;
图3为本发明化合物2的氢谱;
图4为本发明化合物2的质谱;
图5为本发明化合物3氢谱;
图6为本发明化合物3质谱;
图7为本发明化合物4氢谱;
图8为本发明化合物4质谱。
具体实施方式
下面通过具体实施例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
实施例1
本实施例中嘌呤类化合物反应式如下所示:
反应式(ⅰ)中,数字1-12分别代表如下溶剂或催化剂;1为对甲苯磺酸p-tsoh;2为3,4-2h-二氢吡喃;3为四氢呋喃;4为乙二胺;5为n,n-二异丙基乙胺dipea;6为正丙醇;7为生物素biotin;8为n-羟基琥珀酰亚胺nhs;9为碳化二亚胺edci盐酸盐;10为n-乙基二异丙胺dipea;11为n-n二甲基甲酰胺dmf;12为吡啶对甲苯磺酸盐ppts。
(1)化合物1的合成
首先将六氯嘌呤(1g,6.47mmol)、p-tsoh(0.25g,1.29mmol)溶于干燥好的四氢呋喃中,在60℃油浴加热的情况下加入3,4-二氢-2h吡喃(0.885μl,9.700mmol)后,在80℃条件下回流反应12h。反应结束后,利用旋转蒸发仪蒸干溶剂,饱和食盐水和乙酸乙酯萃取,无水硫酸镁干燥萃取后的有机相,用柱层析(hx:ea=4:1)分离提纯后得到化合物1(0.93g,93%)。1hnmr(400mhz,cdcl3)δ8.66(s,1h),8.30(s,1h),5.72(dd,j=10.2,2.7hz,1h),4.11–4.05(m,1h),3.71(td,j=11.7,2.9hz,1h),2.14–1.90(m,3h),1.76–1.52(m,3h).
(2)化合物2的合成
将化合物1(800mg,3.35mmol)溶于正丙醇10ml后,加入乙二胺(316μl,4.73mmol)和dipea(1200μl,6.89mmol)油浴80℃回流反应3小时。利用旋转蒸发仪蒸干溶剂,过硅胶柱(dcm:meoh=5:1)进行粗分离。得到的化合物2(660mg,75.1%)。1hnmr(400mhz,dmso)δ7.72(d,j=3.1hz,1h),7.56(s,1h),7.21(s,1h),5.23(d,j=6.0hz,1h),3.98–3.90(m,2h),3.65(s,2h),3.53–3.46(m,2h),3.04–2.85(m,4h),2.20(t,j=6.4hz,2h),2.05(s,1h),1.84(s,1h).esi-hrmsfor[m+h]+:calcd.for263.16,found263.25.
(3)化合物3的合成
将生物素(559mg,2.29mmol)用干燥好的dmf溶解后,加入nhs(263.mg,2.29mmol)和edci盐酸盐(1096mg,5.72mmol)在室温下反应7h后,向上述反应混合物中加入dipea(1659μl,9.52mmol)和化合物2(500mg,1.91mmol)并继续在常温下反应3h,冷冻干燥去除溶剂后,用柱层析(dcm:meoh=10:1)分离纯化的化合物3(367mg,35.2%)。1hnmr(400mhz,dmso)δ8.41(s,1h),8.29(s,1h),7.84(s,1h),6.49(s,1h),6.43(s,1h),5.70(s,1h),4.35(s,1h),4.16(s,1h),4.05(s,1h),3.73(s,1h),3.58(s,2h),3.33(t,j=10.5hz,3h),3.11(s,1h),2.85(s,1h),2.65–2.58(m,1h),2.35(s,1h),2.11(s,2h),2.00(s,2h),1.79(s,1h),1.63(s,3h),1.53(d,j=7.1hz,3h),1.33(d,j=7.2hz,2h)。esi-hrmsfor[m+h]+:calcd.for511.22,found511.20.
(4)化合物4的合成
将化合物3(200mg,0.41mmol)与ppts(21mg,0.08mmol)溶解于dmf中,常温反应15h,脱去thp保护。冷冻干燥溶剂,用柱层析(dcm:meoh=10:1)分离纯化得到化合物4(120mg,72.5%)。
1hnmr(400mhz,dmso)δ12.92(s,1h),8.18(s,1h),8.09(s,1h),7.95(s,1h),7.61(s,1h),6.42(s,1h),6.36(s,1h),4.32–4.27(m,1h),4.10(dd,j=10.3,5.0hz,1h),3.52(s,1h),3.29(d,j=5.8hz,2h),3.16(s,1h),3.03(s,1h),2.77(s,1h),2.58(s,1h),2.05(t,j=7.4hz,2h),1.63–1.36(m,4h),1.28(dt,j=14.0,7.0hz,2h)。esi-hrmsfor[m+h]+:calcd.for427.16,found427.20.
分别检测mta及合成的化合物与测试蛋白的亲和常数,将mta及实施例1合成的化合物4使用实验者自配的或者微量热泳动技术(microscalethermophoresis,mst)优化的缓冲液(50mmtris(ph7.4),150mmnacl,10mmmgcl2,0.05%tween-20)做16个梯度稀释,一般最高的浓度值为预计kd值的20倍。10μl梯度稀释的合成的化合物和10μl固定浓度的标记蛋白混合后静置反应一会,混合的样品装载到玻璃毛细管中,然后通过mst-nt.115设备进行分析。结果显示其亲和常数相近,分别是347μm与933μm,其亲和常数相近,表示合成的化合物可以替代底物mta进行筛选体系的构建。
以上所述,仅为本发明创造较佳的具体实施方式,但本发明创造的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明创造披露的技术范围内,根据本发明创造的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明创造的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!