核酸样本处理方法、测序方法和试剂盒与流程
[0001]
本发明属于生物大分子检测分析领域,特别地,涉及一种核酸样本处理方法、一种测序方法和一种试剂盒。
背景技术:
[0002]
高通量测序包括二代测序和单分子测序等,在上机检测之前,涉及对核酸样本的处理。特别是基于芯片检测的核酸序列测定,需要对待测核酸(模板)进行处理,以将其连接到芯片上,以实现序列测定。
[0003]
目前市售的基于芯片检测的测序平台,一般都有配套的芯片出售,配套出售的芯片上带有特定序列(探针),模板一般需要通过各种处理以包含该特定序列或者与该特定序列匹配的序列的至少一部分,以使模板能够结合到芯片;所称的各种处理,对于长核酸片段,例如基因组dna,具体地,通常涉及机械或者酶断裂核酸,获得核酸片段,对核酸片段进行修复比如补平其末端和/或使其具有特定粘性末端,以使该核酸片段能够与特定序列(接头)连接,获得测序文库。
[0004]
目前,对核酸的上机前的处理的各个步骤,有各种市售试剂盒,例如,片段化试剂盒,末端修复任选的加da尾试剂盒,接头试剂盒,接头连接试剂盒以及各步骤纯化试剂盒包括检测和/或去除前一反应体系中的干扰后续反应的特定试剂的试剂盒等。而通常地,市售试剂盒都没有披露其具体配方,包括酶的种类、特点、浓度、具体反应溶液配置等。
[0005]
快速简便的上机前核酸的处理方法,包括相关的溶液体系,有待进一步开发和改进。
技术实现要素:
[0006]
本发明提供了一种对dna样本进行处理的方法,该方法中将多种酶混合于同一反应体系中一步实现dna断裂、末端修复和加da尾,简便快捷,节省上机前样本处理的操作时间,利于工业化;同时本发明提供了一种对样本测序的方法,通过探针捕获利用本发明中提供的dna样本进行处理的方法处理的dna样本,课进行二代及三代技术进行测序;另外本发明提供了一种试剂盒,试剂盒中的end prep mix为多种酶和缓冲液混合形成,用于对dna样本进行处理及对样本进行测序可简化操作,简便快捷。
[0007]
本发明一方面提供了一种对dna样本进行处理的方法,该方法包括在第一预混合体系中对所述dna进行片段化、末端修复和加da尾处理,获得第一产物;所述第一预混合体系包括预混合酶,所述预混合酶包括dna片段化酶、多核苷酸激酶和dna聚合酶。
[0008]
可以理解,上述方法中的dna片段化酶的作用使dna样本片段化,达到所需片段的大小,在本发明的实施方案中,dna片段化酶可选自t7脱氧核糖核酸内切酶i、鼠疫杆菌噬菌体phia1122内切核酸酶、噬菌体phiye03-12内切核酸酶、噬菌体t3内切核酸酶、噬菌体t3脱氧核糖核酸内切酶、假单胞菌噬菌体gh-1内切核酸酶、恶臭假单胞菌kt2440脱氧核糖核酸内切酶i和玫瑰噬菌体s101rp内切核酸酶i,可选用的试剂盒为天根dna片段化试剂盒
ng305-02、qiagen cm0162片段化酶试剂盒和neb片段化酶等。
[0009]
在一个示例中,上述方法中的dna不小于5kb,经片段化的第一产物为100~400bp。基于测序仪器测序长度的限制,在一示例中,选择的第一产物的大小为100-300bp。
[0010]
在一个示例中,上述方法中的dna的含量为10ng~60ng;优选的,上述方法中的dna的含量为15ng~50ng,dna的含量的多少与反应体系的大小、所用的酶量的多少以及所需文库量的多少直接相关,dna量太少构建的文库的量太少测序获得数据不能满足要求,同时,如果dna含量太多,在一定的体系内,dna不能被充分片段化及加接头等反应,构建的文库中会出现很多无效片段。选择dna含量为15ng~50ng,是经过多次测试,在32℃的片段化反应温度下,可有效进行反应,同时利用该dna含量构建的文库满足上机测序的需求。
[0011]
在一个示例中,上述第一预混合体系还包括mg
2+
,所述mg
2+
的浓度为60mm~100mm,优选的,上述第一预混合体所述mg
2+
的浓度为60mm~75mm。不同酶对mg
2+
的需求不同,多种酶混合会影响酶反应的最佳条件的选择,在本发明的一个实施例中,第一预混合体系与基因组dna片段化,末端修复和加da尾反应的总体系的体积比为1:5,即在总反应体系中,混合酶反应选用的mg
2+
的浓度为12mm~15mm,选用的mg
2+
的浓度要高于常规酶反应所选用的浓度6mm。
[0012]
在一个示例中,上述方法的第一预混合体系中,dna片段化酶的含量是多核苷酸激酶的至少2倍,所述的含量为总活力单位u,dna聚合酶选自dna聚合酶i和taq dna聚合酶中的至少一种,优选的,dna聚合酶由dna聚合酶i的klenow片段和taq dna聚合酶组成。多核苷酸激酶为t4多核苷酸激酶。
[0013]
在一个示例中,上述第一预混合体系中dna片段化酶、多核苷酸激酶、dna聚合酶i的klenow片段和taq dna聚合酶的量的比例为[25,35]:[6,10]:[1,3]:[3,5],所述的量的比例为总酶活力单位u的比例,此比例是经过多次实验优化而获得的比例,可以同时满足样本对不同酶的需求。在本发明的一个实施例中,dna片段化酶、多核苷酸激酶、dna聚合酶i的klenow片段和taq dna聚合酶的总酶活力单位比例为18:6:1:1,taq酶在第一预混合体系中的浓度为(1/3~2/3)u/ul。其中taq酶活力单位定义为37℃、30分钟内使10nmol的dntp掺入酸不溶性沉淀物所需要的酶量,定义为1个活性单位(u);klenow片段酶活力单位定义为37℃、30分钟内使10nmol的dntp掺入酸不溶性沉淀物所需要的酶量,定义为1个活性单位(u);核苷酸激酶活力单位定义为37℃、30分钟内使1nmol的[γ-32p]atp掺入酸不溶性沉淀物所需要的酶量定义为1个活性单位(u);dna片段化酶活力单位定义为在20ml的反应中,在37℃,在30分钟内,将1μg的超螺旋puc(at)转变为线性形式(两链切开)或切刻形式(单链切开)所需的酶量定义为1个活性单位(u)。
[0014]
上述任一dna样本处理方法,基于测试试验以及优化后的混合酶体系,使多个样本处理模块于同一反应体系中、一步高效地实现,包括dna断裂、末端修复和加da尾,获得的处理后的核酸适合置于各种测序平台上检测,操作简便快捷,节省上机前样本处理所需的时间,利于工业化;进一步地,经过该处理的获得的产物,无需从反应体系中进行分离或纯化,可直接进入下一处理过程。
[0015]
在一个示例中,上述方法还包括在第二预混合体系中使所述第一产物的至少一个末端带有预设序列,获得第二产物,第二预混合体系包括所述第一预混合体系和所述预设序列。
[0016]
进一步,上述第二预混合体系还包括连接酶,如t4 dna连接酶。
[0017]
进一步,上述预设序列为双链dna,由互补的第一链和第二链组成,所述预设序列具有至少一个单链末端。
[0018]
在一示例中,上述第一链的5'末端具有磷酸基团,所述第二链的3'末端不具有羟基,第一链的5'末端与第一产物连接,利用此预设序列进行连接反应,只有第一链与第一产物连接,再不需要pcr的文库构建过程中,利用此方法构建的文库,可以直接用于序列捕获。
[0019]
进一步,80℃≥(tm1-tm2)≥10℃并且90℃≥tm1≥50℃,tm1为所述第一链的溶解温度,tm2为所述第二链的溶解温度。具体的,tm1为71℃,tm2为45.6℃,利用两条链溶解温度的差异降低在序列捕获的过程中第二链对捕获效率的影响。
[0020]
在一示例中,上述预设序列选自seq id no:1和seq id no:2、seq id no:1和seq id no:3和/或seq id no:4和seq id no:5。
[0021]
上述任一方法,基于1-12任一项的方法,通过连接反应和/或聚合反应使待测核酸具有预设序列(本文也称为接头),利用任一项方法中涉及的反应体系以及设计的接头,能够快速且高效的制得包含有特定序列的待测核酸(测序文库),满足后续上机反应的要求。
[0022]
本发明第二方面提供了一种对dna序列进行测定的方法,包括:
[0023]
利用上述dna样本进行处理的方法对dna进行处理,获得第一产物和第二产物;
[0024]
使第二产物连接到固相基质表面上,对第二产物进行测序,以确定所述dna的至少一部分序列。
[0025]
在一示例中,所述第二产物带有光学可检测标记,如荧光分子。
[0026]
在一示例中,上述固相基质表面上具有探针,第二产物通过所述探针连接到所述固相基质表面上,探针的长度为20-80nt。固体基质选自玻璃、塑料和磁珠中的至少一种。
[0027]
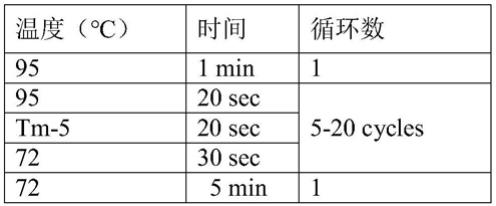
在一示例中,上所述预设序列为双链dna,由互补的第一链和第二链组成,所述预设序列的第一链具有单链末端。进一步,第一链与所述探针完全互补,所述第一链不长于所述探针。进一步,tm2-5℃≤t≤tm1-5℃,t为所述使第二产物连接到固相基质表面上的反应温度,也为探针进行序列捕获的温度为t,tm1为所述第一链的溶解温度,tm2为所述第二链的溶解温度,利用该反应温度可以有效降低第二链对探针进行序列捕获时的影响。
[0028]
在一示例中,上述dna序列进行测定方法还包括在第二产物连接到固相基质表面之前,对第二产物进行变性处理。
[0029]
上述dna序列进行测定方法中,通过设计选定满足特定要求的探针和/或反应条件,使测序文库能够高效地连接到固相基质表面,且稳定地连接,利于稳定高效地测序获得模板信息;同时,这些任一项方法,具有前面任一示例方法的优点和技术特征,在此不再赘述。
[0030]
本发明第三方面提供了一种试剂盒,试剂盒包括第一预混合体系,用于实施上述的dna样本进行处理的方法和dna序列进行测定方法。包含一预混合体系的试剂盒用于样本dna处理时,可以一步实现dna断裂、末端修复和加da尾,简便快捷,节省dna处理时间。
[0031]
在一示例中,试剂盒还包括所述第二预混合体系。
[0032]
在一示例中,试剂盒还包括探针,用于实施上述dna序列进行测定方法,第二产物通过所述探针连接到所述固相基质表面上,探针的长度为20-80nt的dna序列。
[0033]
进一步,探针与第一链互补,探针不短于所述第一链,预设序列为双链dna,由互补
的所述第一链和第二链组成,预设序列具有一个单链末端,单链末端位于所述第一链。
[0034]
该些任一项中的试剂盒,能够用于实现上述任一项的方法,使任一项方法具有前面所说的优点,利于工业化。任一试剂盒,能够用于上机前核酸的处理;可选的,以及用于核酸序列测定。
附图说明
[0035]
图1:实施例2中f4,f5,f6和f7 4个样本加完接头后的产物labchip检测图;
[0036]
图2:实施例2中f4,f5,f6和f7 4个样本pcr产物的labchip检测图;
[0037]
图3:实施例3中n5、n6、n13、n14、n21、n22、n23、n24、o5、o6、o11和o12 12个样本经过打断、末端修复和加a后处理的产物labchip检测图。
具体实施方式
[0038]
除非特殊说明,本文所用术语具有本发明所属领域中的一般含义。
[0039]
下面参考具体实施例,对本发明进行说明,需要说明的是,这些实施例仅仅是说明性的,而不能理解为对本发明的限制。
[0040]
材料和试剂说明
[0041]
接头1(d9-t):由两条单链核酸(第一条链和第二条链)形成的双链核酸,即seq idno:1与seq id no:2形成的双链核酸,或者seq id no:1与seq id no:3形成的双链核酸:
[0042]
第一条链:5
’-
agatgtgtataagagacagt-3’(seq id no:1)
[0043]
第二条链:5
’-
actgtctcttatacacatctgagtggaactggatggtcgcaggtatcaaggatt-3’(seq id no:2)或者5
’-
ctgtctcttatacacatctgagtggaactggatggtcgcaggtatcaagga-3'(seq id no:3)
[0044]
接头2:由两条单链核酸(第一条链和第二条链)形成的双链核酸,例如,与部分illumina测序平台匹配的p5和/或p7接头;可购自市售试剂盒,例如诺唯赞:
[0045]
第一条链:5'-acactctttccctacacgacgctcttccgatc-s-t-3'(seq id no:4)
[0046]
第二条链:5'-p-gatcggaagagcacacgtctgaactccagtc-3'(seq id no:5)
[0047]
(-s-表示硫代,-p表示磷酸化)
[0048]
实施例中未注明具体技术或条件的,均按照常规实验条件,或按照制造厂商说明书建议的条件。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0049]
实施例1
[0050]
以下示例制备试剂盒的方法。
[0051]
试剂盒包含管1,管1为具有末端修复功能的混合体系(也称为end prep mix)。在一种情况中,管1是包含多核苷酸激酶和多种dna聚合酶的溶液。在另一种情况中,管1是包含dna片段化酶、多核苷酸激酶和多种dna聚合酶的溶液。
[0052]
dna聚合酶可以补平片段化后的核酸片段的末端,这里,较佳的,所选择的dna聚合酶具有扩增功能,同时具有修复、校对功能,例如选自taq dna聚合酶(简称为taq酶)和klenow片段中的一种或两种;多核苷酸激酶可选择t4多核苷酸激酶(t4 polynucleotide kinase)。
[0053]
在一种情况中,所称的dna聚合酶为taq酶和klenow片段的混合,taq酶、klenow片
段和多核苷酸激酶在管1中酶活力单位(u)比为1:1:6,在管1中taq酶的浓度为(1~2)u/ul。这里,各酶的活力单位的定义如下:taq酶活力单位:37℃、30分钟内使10nmol的dntp掺入酸不溶性沉淀物所需要的酶量,定义为1个活性单位(u);klenow片段活力单位:37℃、30分钟内使10nmol的dntp掺入酸不溶性沉淀物所需要的酶量,定义为1个活性单位(u);核苷酸激酶活力单位:37℃、30分钟内使1nmol的[γ-32p]atp掺入酸不溶性沉淀物所需要的酶量定义为1个活性单位(u)。
[0054]
在一种情况中,管1还包含dna片段化酶,先配置除了dna片段化酶的混合酶液a,再将dna片段化酶溶液加入混合酶液a,制得管1,例如dna片段化酶溶液和混合酶液a以体积比1:2混合,使得dna片段化酶,klenow片段的混合,taq酶、klenow片段和多核苷酸激酶在管1中酶活力单位(u)比为18:1:1:6,管1中taq酶的浓度为混合酶液a中taq酶的浓度的2/3;dna片段化酶可选自天根dna片段化试剂盒ng305-02、qiagen cm0162片段化酶试剂盒或者neb片段化酶。混合酶液a中的taq酶、klenow和多核苷酸激酶可选自neb公司、天根、诺唯赞(vazyme)等公司出售的相应产品。dna片段化酶活力单位:在20ml的反应中,在37℃,在30分钟内,将1μg的超螺旋puc(at)转变为线性形式(两链切开)或切刻形式(单链切开)所需的酶量。
[0055]
在配置该管1时,可分别配置或购买各单独酶溶液,再将各单独酶溶液混合;也可分别配置混合酶液和缓冲液,再将二者混合,例如二者按照体积比1:1混合,以制备得管1,。
[0056]
在一种情况中,在制备管1时,选择购买各商家的单独酶溶液,包括单独的dna片段化酶、t4多核苷酸激酶、taq dna聚合酶和klenow片段试剂盒;再将各市售试剂盒中的单独酶溶液按特定比例混合,以及另外加入mgcl2溶液,例如额外加入不少于60mm的mgcl2,从而制得管1。
[0057]
在一种情况中,管1中的各种酶共同置于下述溶液体系中:10x buffer具体成分为700mm tris-hcl ph 7.6,120-150mm mgcl2,50mm dtt和1mm dntps;这里的1mm dntps指的是每一种dntp的浓度均为1mm。管1中的各种酶的总体积与10x buffer的体积比为1:1,此时获得的管1的混合体系为5x end prep mix,即在利用管1混合体系进行dna片段化,末端修复和加da尾的反应时,管1的混合体系与总反应体系的体积比为1:5;管1中buffer具体成分及浓度分别为:350mm tris-hcl ph 7.6,60-75mm mgcl2,25mm dtt和0.5mm dntps,管1中taq酶的浓度为(1/3~2/3)u/ul。
[0058]
在一些情况中,试剂盒还包含管2,管2为具有连接功能的混合体系(ligation mix),ligation mix由dna连接酶和连接酶缓冲体系组成。dna连接酶可以为blunt/ta ligase,例如t4 dna连接酶。在一些情况中,试剂盒还包含管2,管2为具有连接功能的混合体系(ligation mix),ligation mix由dna连接酶和连接酶缓冲体系组成。dna连接酶可以为blunt/ta ligase,例如t4 dna连接酶。2x ligation mix各物质的浓度为:t4 dna ligase buffer:80-100mm tris-hcl ph 7.8,20mm mgcl2,20mm dtt,10mm atp,50ug/ml bsa,t4 dna ligase的活力单位浓度为,60u~100u/ul。可根据需要配置不同浓度的ligation mix。t4 dna ligase活力单位定义为在20ul反应体系50mm tris-hcl ph 7.5,10mm mgcl2,10mmdtt,5mm atp,25ug/ml bsa中,16℃、30min内,将6ug的λdna hindiii酶切产物的50%连接需要的酶量。ligation mix可选自neb公司货号m0367s试剂盒中的试剂。
[0059]
在一些情况中,试剂盒还包含管3、管4和管5等,例如包含pcr反应试剂的管3、包含
接头的管4和包含引物的管5等。例如,包含独立管装的可用于三代测序文库构建的接头1(d9-t),包含独立管装的适用于二代测序文库构建例如illumina平台的接头2;pcr扩增试剂可选择普通pcr扩增试剂盒(含taq酶)或高保真pcr扩增试剂盒(含高保真酶,如pfu),可选自赛默飞、莫克、beckman等公司的pcr扩增试剂盒。
[0060]
利用该试剂盒,能够高效地构建出适于各种测序平台的文库,包括但不限于二代测序平台和三代测序平台。
[0061]
实施例2
[0062]
本实施例提供了实施例1中的试剂盒在文库构建中的应用。本实施例中共做4个平行实验,所用的样本分别为f4,f5,f6和f7基因组,基因组dna在片段前需要对基因组的质量做初步评估,保证基因组存在于无金属离子螯合剂或其他盐的溶剂中,并且通过凝胶电泳检测基因组完整度。文库构建的具体步骤如下:
[0063]
1.基因组dna片段化,末端修复和加da尾
[0064]
这一步骤将dna进行片段化,同时将片段化dna末端补平,并在5’端进行磷酸化和3’端加da尾。
[0065]
按照表1所示在pcr管中配置反应体系:
[0066]
表1
[0067]
h2o14ulend prep mix4uldna(25ng/ul)2ultotal20ul
[0068]
将pcr管置于pcr仪中,按照如表2所示设置反应条件:
[0069]
表2
[0070]
temptime热盖105℃on32℃15-20min65℃10min4℃hold
[0071]
反应结束后立即进入下一步操作。
[0072]
2.接头连接
[0073]
步骤1反应结束后,在pcr管中按照表3所示配制接头连接体系:
[0074]
表3
[0075]
组分体积步骤1产物20ul接头1(浓度为15um)5ulligation mix(neb m0367s)25ultotal50ul
[0076]
反应条件:室温放置15min。
[0077]
3.纯化
[0078]
使用vahts dna clean beads对反应产物进行纯化,纯化步骤如下所示:
[0079]
1)将接头连接后的体系转移至1.5ml ep管中,加入0.8
×
(40μl)磁珠,吹打混匀10次,室温放置3min。
[0080]
2)将1.5ml ep管放置在磁力架上,静置2-3min,移去上清。
[0081]
3)采用新鲜配置的200μl 80%乙醇进行磁珠上架洗涤。
[0082]
4)移除乙醇后,瞬时离心,再放在磁力架上,用10μl枪去除干净。
[0083]
5)开盖干燥磁珠约5-10min至乙醇完全挥发。
[0084]
6)加入22μlddh2o下架洗脱,充分混匀后室温静置3min,置于磁力架上3min,回收产物20μl,再加入1.2x(24ul)磁珠,吹打混匀10次,室温放置3min。
[0085]
7)将1.5ml ep管放置在磁力架上,静置2-3min,移去上清。
[0086]
8)采用新鲜配置的200μl 80%乙醇进行磁珠上架洗涤。
[0087]
9)移除乙醇后,瞬时离心,再放在磁力架上,用10μl枪去除干净。
[0088]
10)开盖干燥磁珠约5-10min至乙醇完全挥发。
[0089]
11)加入12μlddh2o下架洗脱,充分混匀后室温静置3min,置于磁力架上3min,回收产物13μl。
[0090]
取回收产物1μl(连接产物)进行labchip检测,检测结果见图1,图1显示4个样本加完接头后的主带在320bp。从4个样本的检测结果可看出用end prep mix可以降基因组稳定打断至特定大小的片段。
[0091]
4.文库扩增
[0092]
将回收产物进行pcr扩增。pcr扩增反应体系按照表4所示配制:
[0093]
表4
[0094]
组分体积扩增模块12.5ul连接产物11.5ul正向引物(10um)0.5ul反向引物(10um)0.5ul总计25ul
[0095]
正向引物:5'-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct-3'(seq id no:6)
[0096]
反向引物:5'-gtgactggagttcagacgtgtgctcttccgatct-3'(seq id no:7)
[0097]
pcr扩增反应条件如表5所示:
[0098]
表5
[0099]
[0100][0101]
5.纯化
[0102]
使用vahts dna clean beads对反应产物进行纯化:
[0103]
(1)磁珠平衡至室温后,涡旋振荡混匀vahts dna clean beads。
[0104]
(2)将连接后的pcr体系(25ul)转移至1.5ml ep管中,加入1.8
×
(45ul)磁珠,吹打混匀10次,室温放置3min。
[0105]
(3)将1.5ml ep管放置在磁力架上,静置2-3min,移去上清。
[0106]
(4)采用新鲜配置的200ul 80%乙醇进行磁珠上架洗涤。
[0107]
(5)重复步骤4
[0108]
(6)移除乙醇后,瞬时离心,再放在磁力架上,用10ul枪去除干净。
[0109]
(7)开盖干燥磁珠约5-10min至乙醇完全挥发。
[0110]
(8)将1.5ml从磁力架中取出,进行洗脱:加入22ul ddh2o,涡旋振荡或使用移液器轻轻吹打混匀,于室温静置3min,将ep管短暂离心并置于磁力架中静置,待溶液澄清后,
[0111]
移取20ul上清至新的1.5ml ep管中。
[0112]
取pcr扩增后纯化纯化产物1ul进行labchip检测,检测结果见图2,图2显示4个样本加完接头进行pcr扩增后的产物的主带在320bp。因为利用的引物与接头中序列互补配,对从扩增结果可看出,片段得到有效扩增,所以利用ligation mix可有效的添加接头,4个样本打断加接头后片段大小一致。
[0113]
6.定量上机测序
[0114]
定量合格的文库用于illumia测序仪进行测序。
[0115]
以上相关步骤,较佳地,模板dna量范围为15ng-50ng,od260/od280=1.8-2.0。打断范围为150-200bp
[0116]
推荐接头adapter:input dna摩尔比在100:1-600:1之间。adapter投入量过高可能会导致adapter或adapter dimer残留;投入量不足又会影响连接效率进而导致文库产出降低。adapter工作浓度25um,模板摩尔数(pmol)≈input dna质量(ng)/[0.66*input dna平均长度(bp)]。
[0117]
磁珠用量直接影响可纯化的dna长度下限。乘数越高,可纯化的dna长度下限越短;反之,则越长。例如:1x磁珠只能高效纯化长于250bp的dna,更短的dna会在纯化过程中大量丢失;提高至1.8x后,150bp的dna也可进行高效纯化。
[0118]
洗脱产物可于4℃稳定保存一周;长期保存时应置于-20℃,避免不必要的反复冻融。
[0119]
实施例3
[0120]
本实施例提供了实施例1中的试剂盒在文库构建中的应用2。本实施例中共做12个平行实验,所用的样本分别为n5、n6、n13、n14、n21、n22、n23、n24、o5、o6、o11和o12基因组,基因组dna在片段前需要对基因组的质量做初步评估,保证基因组存在于无金属离子螯合剂或其他盐的溶剂中,并且通过凝胶电泳检测基因组完整度。文库构建的具体步骤如下:
[0121]
1.dna片段化,末端修复和加da尾
[0122]
这一步骤将dna进行片段化,同时将片段化dna末端补平,并在5’端进行磷酸化和3’端加da尾。
[0123]
不同样本反应体系相同,如表6所示,32℃反应时间不同,分别为20min,22min,25min,30min,35min,40min。
[0124]
样本种类及对应的反应时间如下:
[0125]
n5:50ng,32℃20min;n6:50ng,32℃20min;
[0126]
n13:50ng,32℃22min;n14:50ng,32℃22min;
[0127]
n21:50ng,32℃25min;n22:50ng,32℃25min;
[0128]
n23:50ng,32℃35min;n24:50ng,32℃35min;
[0129]
o5:50ng,32℃30min;o6:50ng,32℃30min;
[0130]
o11:50ng,32℃40min;o12:50ng,32℃40min。
[0131]
表6
[0132]
h2o14ulend prep mix4uldna(25ng/ul)2ultotal20ul
[0133]
将pcr管置于pcr仪中,按照如表7所示设置反应条件::
[0134]
表7
[0135]
temp(温度)time(时间)热盖105℃on32℃(20,22,25,30,35,40)min65℃10min4℃hold
[0136]
反应结束后立即进入下一步操作。
[0137]
反应结束后的产物利用labchip进行检测,检测结果见图3所示,检测结果曲线图中主峰最高-最低依此对应的样本为n5、n6、n13、n14、n21、n22、n23、n24、o5、o6、o11和o12。从图3可知,打断时间20min-40min,打断温度为32℃,片段化酶对基因组dna进行有效打断,打断后的片段主带在280bp左右,不同的打断时间对基因组打断片段打断大小影响不大,大小波动在20bp左右。
[0138]
2.接头连接
[0139]
步骤1反应结束后,在pcr管中按照表8所示配制接头连接体系:
[0140]
表8
[0141][0142]
反应条件:室温放置15min。
[0143]
3.纯化
[0144]
使用vahts dna clean beads对反应产物进行纯化,纯化步骤如下所示:
[0145]
12)将接头连接后的体系转移至1.5ml ep管中,加入0.8
×
(40μl)磁珠,吹打混匀10次,室温放置3min。
[0146]
13)将1.5ml ep管放置在磁力架上,静置2-3min,移去上清。
[0147]
14)采用新鲜配置的200μl 80%乙醇进行磁珠上架洗涤。
[0148]
15)移除乙醇后,瞬时离心,再放在磁力架上,用10μl枪去除干净。
[0149]
16)开盖干燥磁珠约5-10min至乙醇完全挥发。
[0150]
17)加入22μlddh2o下架洗脱,充分混匀后室温静置3min,置于磁力架上3min,回收产物20μl,再加入1.2x(24ul)磁珠,吹打混匀10次,室温放置3min。
[0151]
18)将1.5ml ep管放置在磁力架上,静置2-3min,移去上清。
[0152]
19)采用新鲜配置的200μl 80%乙醇进行磁珠上架洗涤。
[0153]
20)移除乙醇后,瞬时离心,再放在磁力架上,用10μl枪去除干净。
[0154]
21)开盖干燥磁珠约5-10min至乙醇完全挥发。
[0155]
22)加入12μlddh2o下架洗脱,充分混匀后室温静置3min,置于磁力架上3min,回收产物10μl。
[0156]
回收产物可于4℃稳定保存一周。长期保存置于-20℃,避免不必要的反复冻融。
[0157]
4.序列捕获,上机测序
[0158]
定量合格的文库用于序列捕获,并利用单分子测序仪进行测序。
[0159]
利用公开专利申请cn201510501968.7说明书中公开的方法在芯片上固定探针(seq idno:8),制备的文库用3*ssc杂交液稀释,然后与芯片上固定的探针进行杂交。然后根据cy3的信号来判断接头序列与探针杂交的数目。
[0160]
seq id no:8:5
’-
tttttttttttccttgatacctgcgaccatccagttccactcagatgtgtataagagacag-3’。
[0161]
文库芯片杂交的流程如下:
[0162]
(1)芯片选择:所用的芯片的基地玻璃为schott公司的环氧基修饰的玻璃芯片,通过探针上的氨基和芯片表面的环氧基团反应的方法,例如,可参看公开专利申请号cn201811191589.2披露的方法来固定seq id no.4所示的探针,在110
×
110μm区域内固定的探针密度约为18000dot/fov即在110
×
110μm视野内有18000个亮点。
[0163]
(2)杂交液配置:杂交液配置体系如表3所示,所用缓冲液为20
×
ssc缓冲液(西格玛,#s6639-1l),终浓度为成3
×
ssc,文库的终浓度为1nm,总体积为40μl。将配置好的杂交液95℃变性2min,迅速至冰上冷却。
[0164]
表3
[0165]
20
×
ssc缓冲液6μl文库终浓度为1nm去核酸水补足至40μl
[0166]
(3)将上述变性后的杂交液迅速加载至芯片上,再将芯片放置55℃30min,使文库与芯片表面的探针进行杂交。
[0167]
(4)依次用3
×
ssc、1
×
ssc、0.1
×
ssc冲洗芯片。
[0168]
利用genocare第三代测序平台对杂交捕获的文库进行测序.
[0169]
以上示意较佳实施例,并非用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。尽管上面已经示出和描述了本发明的实施方式,可以理解的是,上述实施方式是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施方式进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1