限制几何构型茂金属催化剂及其制备方法与应用与流程
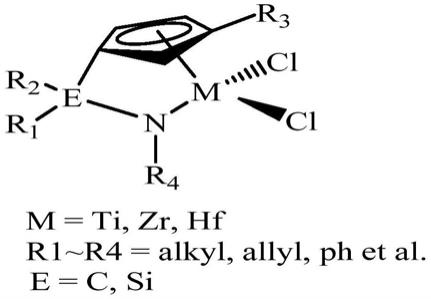
1.本发明涉及一种吡咯茂杂环限制几何构型茂金属催化剂及其制备方法,以及使用其制备聚烯烃的方法。
背景技术:
2.当配体上的桥联杂原子与金属原子一起形成半夹心结构的双官能团的化合物,在mao的作用下它们表现出较高催化烯烃聚合的能力,人们称之为限制几何构型催化剂(cgc)。限制几何构型催化剂较其它茂金属催化剂的特点是离域π电子键合基团cp*上引入限制构型的取代基团,从而使配合物中金属原子的几何构型受到限制,cp*-金属中心原子-与桥基相连的配位基团构成的夹角与类似的配合物相比较小,因此,限制几何构型催化剂中金属原子具有更“open”的特点。
3.1990年dow化学公司合成了所谓的“限定构型催化剂”。它是用胺基取代si1或c1桥联茂金属催化剂结构中的一个环戊二烯(或茚基、芴基),是一种单环戊二烯与第ⅳ副族过渡金属以配位键形成的络合物,单环戊二烯基、过渡金属与杂原子(例如氮)间的键角小于115
°
,强lewis酸体系可将该催化剂活化成高效正离子。一方面二齿配位体稳定了金属电子云,另一方面短桥基团的存在又使配位体的位置发生偏移,从空间构型上使催化剂活性中心只能向一个方向打开,从而达到限定几何构型的目的。所以dow化学公司称其为限制构型催化剂(constrained-geometry catalyst),即cgc。典型结构如式1所示。
[0004][0005]
式1 cgc典型结构
[0006]
shapiro and bercaw等人合成的第一种cgc使用叔丁基作为给电子体,随后不久,实验人员使用其它基团试图改性,如更强吸电子性的苯环和金刚烷等等。
[0007][0008]
式2第一个限制构型催化剂
[0009][0010]
式3苯环和金刚烷取代的限制构型催化剂
[0011]
除了烷基和芳基以外,研究人员也引入其他基团,如磺酰氨基,吡咯基,肼基和亚氨基等等,与烷基和芳基相比,磺酰氨基、吡咯基等有更强的吸电子性,所以ti-n键更长,也有利于α-烯烃的插入。
[0012][0013]
式4其它取代基团的限制构型催化剂
[0014]
当然,除了n基团作为给电子基团外,还有p,s,o等杂原子的基团也作为给电子体,但相比于含n给电子体,其它杂原子做给电子体合成的cgc不论活性还是所得到的聚合物性能都不理想,因此,目前来说,其它杂原子做给电子体的cgc研究很少,比如,关于含s原子的cgc目前只报道了一例,而且基本没有催化活性。
[0015][0016]
式5其它杂原子做给电子基团的限制构型催化剂
[0017]
由于cgc催化剂独特的立体结构可以允许各种烯烃单体插入,它既可以催化烯烃均聚,也可以催化乙烯/α-烯烃进行共聚。自从第一个cgc被合成以来,已经接近30年了,一系列的cgc被合成出来,但大部分cgc结构差异不大,催化聚合的聚合物性能也没有突破进展,并且由于dow公司对硅桥限制几何构型催化剂的专利保护,我们国家在生产低密度聚乙烯(lldpe)和poe等聚烯烃产品受到许多限制,所以开发结构新颖的限制几何构型催化剂则显得非常有意义。
技术实现要素:
[0018]
本发明的主要目的在于提供一种限制几何构型茂金属催化剂及其制备方法与应用,以能够灵活调控用于催化烯烃聚合时的活性以及α-烯烃单体的插入率。
[0019]
为了实现上述目的,本发明提供了一种限制几何构型茂金属催化剂,该限制几何构型茂金属催化剂具有如下式ⅰ结构:
[0020][0021]
其中,r1、r2和r3分别独立选自h、ch
3-、饱和的或含有双键的直链或支链c
2-c
10
烃基中的一种;r4为直链或支链的c
1-c5烷基。
[0022]
本发明所述的限制几何构型茂金属催化剂,其中,所述r4为ch
3-、ch3ch
2-、ch3ch2ch
2-和ch3ch2ch2ch
2-所组成群组中的一种。
[0023]
为了达到上述目的,本发明还提供了一种限制几何构型茂金属催化剂的制备方法,该制备方法包括如下步骤:
[0024]
步骤1,使式ⅱ2-吡咯甲醛或其衍生物与茚发生反应,制备式ⅲ含氮富烯,r1、r2和r3分别独立选自h、ch
3-、饱和的或含有双键的直链或支链c
2-c
10
烃基中的一种;
[0025]
步骤2,还原式ⅲ含氮富烯,生成式ⅳ含吡咯n杂环的配体;以及
[0026]
步骤3,式ⅳ含吡咯n杂环的配体与ti[n(r4)2]4发生络合反应,生成式ⅰ茂金属催化剂,r4为直链或支链的c
1-c5烷基;
[0027][0028]
本发明所述的限制几何构型茂金属催化剂的制备方法,其中,所述r4为ch
3-、ch3ch
2-、ch3ch2ch
2-和ch3ch2ch2ch
2-所组成群组中的一种。
[0029]
本发明所述的限制几何构型茂金属催化剂的制备方法,其中,所述步骤1为:将式ⅱ2-吡咯甲醛或其衍生物与茚溶于有机溶剂中,降温至-10~5℃,滴加四氢吡咯,然后升至室温搅拌反应0.5-20小时,得到式ⅲ含氮富烯;
[0030]
其中,式ⅱ2-吡咯甲醛或其衍生物、茚与四氢吡咯三者物质的量之比为1:1~5:1~5。
[0031]
本发明所述的限制几何构型茂金属催化剂的制备方法,其中,所述步骤2为:将步骤1得到的式ⅲ含氮富烯溶解于有机溶剂中,降温至-10~5℃,滴加溶于有机溶剂中的氢化铝锂,然后升温至40℃~70℃,搅拌反应5~50h,得到式ⅳ含吡咯n杂环的配体;
[0032]
其中,式ⅲ含氮富烯与氢化铝锂的摩尔比为1:0.5~5。
[0033]
本发明所述的限制几何构型茂金属催化剂的制备方法,其中,所述步骤3为:将步骤2得到的式ⅳ含吡咯n杂环的配体溶于有机溶剂中,降温至-10~5℃,然后滴加溶于有机溶剂的ti[n(r4)2]4,升温至50℃~100℃,搅拌反应1h~30h,得到茂金属催化剂;
[0034]
其中,式ⅳ含吡咯n杂环的配体与ti[n(r4)2]4的摩尔比为1:0.5~5。
[0035]
本发明所述的限制几何构型茂金属催化剂的制备方法,其中,所述步骤1、步骤2和步骤3皆在惰性气体保护下进行,所述惰性气体为氮气、氦气和氩气所组成群组中的一种或几种。
[0036]
为了达到上述目的,本发明尚提供了一种烯烃聚合反应方法,其在上述的限制几何构型茂金属催化剂的作用下进行。
[0037]
本发明所述的烯烃聚合反应方法,其中,所述烯选自乙烯、丙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯、1-癸烯、1-十一碳烯、1-十二碳烯、1-十四碳烯、1-十六碳烯、1-二十碳烯、二环戊二烯、1,4-丁二烯、1,5-戊二烯、1,6-己二烯、苯乙烯、α-甲基苯乙烯和二乙烯基苯所组成群组中的至少一种。
[0038]
本发明所述的烯烃聚合反应方法,其中,上述的限制几何构型茂金属催化剂为主催化剂,烷基铝或者铝氧烷化合物为助催化剂,主催化剂与助催化剂的摩尔比为1:500-2000。
[0039]
本发明催化剂用吡咯基杂环取代传统上的采用的叔丁基为给电子体,通过步骤少的胺消除法制备催化剂,通过吡咯基上取代基位置的不同,控制金属中心的电子环境与空间环境,进而调控制备聚合物的活性以及α-烯烃单体的插入率,与叔丁胺基相比,吡咯基有更强的吸电子性,所以ti-n键更长,也有利于α-烯烃的插入,预期得到更高的单体插入率,制得性能优异的聚合物,而且本发明所得催化剂具有合成路线短,合成工艺简单,工业成本低廉的优点。
具体实施方式
[0040]
以下对本发明的实施例作详细说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
[0041]
本发明提供了一种限制几何构型茂金属催化剂,该限制几何构型茂金属催化剂具有如下式ⅰ结构:
[0042][0043]
其中r1、r2、r3、r4结构分别为:
[0044]
r1为h、ch
3-、c
2-c
10
的饱和的或含有双键的直链烃基或支链烃基中的一种;
[0045]
r2为h、ch
3-、c
2-c
10
的饱和的或含有双键的直链烃基或支链烃基中的一种;
[0046]
r3为h、ch
3-、c
2-c
10
的饱和的或含有双键的直链烃基或支链烃基中的一种;
[0047]
r4为直链或支链的c
1-c5烷基,优选为ch
3-、ch3ch
2-、ch3ch2ch
2-或ch3ch2ch2ch
2-中的一种,进一步优选为ch
3-或ch3ch
2-。
[0048]
本发明还提供了一种限制几何构型茂金属催化剂的制备方法,该制备方法包括如下步骤:
[0049]
步骤1,使式ⅱ2-吡咯甲醛或其衍生物与茚发生反应,制备式ⅲ含氮富烯,r1、r2和r3分别独立选自h、ch
3-、饱和的或含有双键的直链或支链c
2-c
10
烃基中的一种;
[0050]
步骤2,还原式ⅲ含氮富烯,生成式ⅳ含吡咯n杂环的配体;以及
[0051]
步骤3,式ⅳ含吡咯n杂环的配体与ti[n(r4)2]4发生络合反应,生成式ⅰ茂金属催化剂,r4为直链或支链的c
1-c5烷基;
[0052][0053]
具体而言,步骤1为:在schlenk瓶中将式ⅱ2-吡咯甲醛或其衍生物和茚溶于有机溶剂中,并降温至-10~5℃,例如可以将schlenk瓶置于冰水浴中,然后缓慢滴加四氢吡咯,滴加完毕后升温至室温,搅拌0.5-20小时,优选搅拌5-15小时;再用有机溶剂分液萃取、洗涤后取有机相,将有机相干燥、旋蒸得到式ⅲ含氮富烯。2-吡咯甲醛或其衍生物、茚与四氢吡咯三者物质的量之比为1:1-5:1-5,优选的2-吡咯甲醛或其衍生物、茚与四氢吡咯三者物质的量之比为1:1-3:1-3。其中,有机溶剂优选为甲醇、乙醇、甲醛、乙醛、乙醚、甲苯、乙苯中的一种。
[0054]
步骤2为:在schlenk瓶中将步骤1制备的含氮富烯溶解在有机溶剂中,例如为四氢呋喃中,并降温至-10~5℃,例如将schlenk瓶置于冰水浴中,将溶有氢化铝锂的四氢呋喃溶液缓慢加入反应瓶中,后撤去冰水浴,缓慢加热到40℃-70℃,恒温搅拌反应5-50h,加入有机溶剂,分液、萃取、洗涤、干燥有机相后旋蒸得到配体。其中,式ⅲ含氮富烯与氢化铝锂的摩尔比为1:0.5-5;萃取洗涤用有机溶剂为乙醇、乙醛、乙醚、甲苯、乙苯中的一种。
[0055]
步骤3:在schlenk瓶中将ti(n(r4)2)4溶于有机溶剂并并降温至-10~5℃,例如置于冰水浴中,将步骤2制备的配体溶于有机溶剂后缓慢加入到schlenk瓶中,撤去冰水浴加热到50℃-100℃,恒温搅拌反应1h-30h,反应结束之后抽干溶剂,最后在有机溶剂中低温重结晶得到产物。其中,有机溶剂为甲烷、乙烷、丙烷、乙醇、乙醛、乙醚、甲苯、乙苯中的一种或几种。
[0056]
本发明提供的吡咯茂杂环限制几何构型茂金属催化剂的制备方法,整个反应过程中一直在惰性气体保护下进行,所述的惰性气体为氮气、氦气、氩气中的一种。
[0057]
本发明尚提供了一种烯烃聚合反应方法,其在上述的限制几何构型茂金属催化剂的作用下进行。优选的,上述的限制几何构型茂金属催化剂作为主催化剂,烷基铝或者铝氧
烷化合物作为助催化剂,对烯烃聚合反应进行催化。其中,烷基铝或者铝氧烷化合物为甲基铝氧烷、三甲基铝、三乙基铝、三异丁基铝或它们的混合物,主催化剂与助催化剂的物质的量的比为1:500-2000,优选为1:600-1500。
[0058]
本发明限制几何构型茂金属催化剂可以用于催化聚合的烯烃单体包括乙烯、丙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯、1-癸烯、1-十一碳烯、1-十二碳烯、1-十四碳烯、1-十六碳烯、1-二十碳烯、二环戊二烯、1,4-丁二烯、1,5-戊二烯、1,6-己二烯、苯乙烯、α-甲基苯乙烯以及二乙烯基苯中的至少一种。
[0059]
其中,烯烃聚合反应可以按照以下方法进行:
[0060]
乙烯均聚、乙烯与α-烯烃的共聚反应在一套高压反应装置中进行,高压釜容量为250ml,可以实时监控和调节温度与转速。在反应之前,要对反应釜进行真空预热,当达到设定的温度之后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中称量好主催化剂,将主催化剂溶解在甲苯溶剂中,依次往加料斗中加入一半溶剂、共聚单体1-己烯或1-辛烯、助催化剂mao、主催化剂,最后用剩余一半溶剂冲洗,将加料斗中残留的催化剂冲进加料器中,关闭加料器阀门,拿出手套箱,用氩气将加料器中溶液冲进反应釜中。打开阀门,将乙烯钢瓶压力升至设定值,打开搅拌,开始计时,在反应过程中控制温度尽量在设定值左右,反应结束后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(30ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,计算催化剂催化活性。
[0061]
该类催化剂在催化乙烯均聚时,以mao为助催化剂,催化活性高达4.5
×
106g/(mol
·
zr
·
h);催化乙烯/1-己烯共聚时,以mao为助催化剂,催化活性达到2.3
×
106g/(mol
·
zr
·
h),1-己烯插入率9.81%;催化乙烯/1-辛烯共聚时,以mao为助催化剂,催化活性达到1.3
×
106g/(mol
·
zr
·
h),1-辛烯插入率8.84%;而对比一般的叔丁胺基碳桥限制几何构型催化剂,如[t-bun(me)2c(η
5-c5h4)]zrcl2,相同条件下,催化所得乙烯/1-己烯共聚物的插入率只有8.34%,乙烯/1-辛烯插入率只有5.46%,而催化活性在同一数量级,所以本发明所得催化剂更有工业应用价值。
[0062]
综上所述,本发明采用胺消除法合成含吡咯n杂环的第ⅳb族限制几何构型催化剂,简化了合成步骤,在合成富烯和配体时反应产率均达到了90%以上,而且在合成催化剂这一步产率也大大提高,因此,总体而言,通过这种方法合成的催化剂收率提高了很多,研究发现,与烷基-n给电子体相比,吡咯基的吸电子性更强,所以zr-n键更长,使得茚基、金属中心原子和吡咯基之间夹角比一般烷基n给电子体cgc要小,更有利于α-烯烃的进攻,这种类型的催化剂共聚活性会更好,而通过改变吡咯基上取代基的位置,影响活性中心的空间结构和电子环境,使其催化行为产生差异,有效提高催化活性和共聚物中α-烯烃的插入率。
[0063]
而且,本发明提供的催化剂合成路线简单,催化剂收率高,步骤少,用于催化乙烯与α-烯烃共聚合时活性好,α-烯烃单体插入率高,拥有较好的应用价值。
[0064]
以下将通过具体实施例对本发明技术方案进行进一步说明。
[0065]
实施例1
[0066]
络合物1[(η
5-c9h6)ch2(α-c4h3n)]ti(nme2)2的合成
[0067]
(1)富烯(c9h6)ch(α-c4h3nh)的制备
[0068][0069]
在200ml schlenk瓶中将2-吡咯甲醛(4g,42mmol)和新蒸的茚(12.3g,106mmol)溶于40ml甲醇,在冰水浴下缓慢滴加四氢吡咯(6g,84mmol),在滴加过程中可以看到溶液逐渐由浅黄色变为亮黄色,表明已经开始反应,滴加完后自然升至室温,搅拌90min。在冰水浴下加50ml蒸馏水淬灭,再加冰醋酸(5g,84mmol)调至ph=7,加入20ml乙醚,用分液漏斗分液,取上层有机相,水相用乙醚萃取三次(30ml
×
3),合并有机相,再用饱和食盐水洗涤两次,将得到的有机相放入锥形瓶中用无水mgso4干燥6h。旋蒸得到土黄色固体,再用柱层析分离(淋洗剂:乙酸乙酯/石油醚=1/10)得到纯净的产物6.97g(40mmol),收率为86%。
[0070]
通过1h nmr,
13
c nmr,确认其化学结构。(1h nmr(cdcl3,25℃):8.32(br s,1h,nh),6.77(s,1h,ch),7.48(m,1h,c4h3nh),7.23(m,1h,c4h3nh),7.06(m,1h,c4h3nh),7.11(m,2h,c
6-h),6.87(m,2h,c
6-h),6.56
–
6.22(m,2h,cp-h);
13
c nmr(100mhz,cdcl3):δ140.21,136.70,132.61,131.68,129.10,125.47,124.04,123.77,121.21,120.03,117.58,117.12,113.68,110.28.)
[0071]
(2)配体(c9h7)ch2(α-c4h3nh)的制备
[0072][0073]
在200ml schlenk瓶中将7.33g富烯(c9h6)ch(α-c4h3nh)(40mmol)溶解在100ml thf中,在冰水浴下将38ml的lialh4/thf(1m)缓慢加入反应瓶中,可以看到有很多气泡产生,表明反应正在进行,控制滴加速度至大约半小时加完,撤去冰水浴,缓慢加热到50℃,反应15h,在冰水浴下用50ml水淬灭,布氏漏斗过滤除去大量铝盐,加入20ml乙醚,用分液漏斗分液,取上层有机相,水相用乙醚萃取三次(30ml
×
3),合并有机相,再用饱和食盐水洗涤两次,将得到的有机相放入锥形瓶中用无水mgso4干燥6h。旋蒸得到白色固体,再用柱层析分离(淋洗剂:乙酸乙酯/石油醚=1/20)得到纯净的产物5.91g(30mmol),收率为79%。
[0074]
通过1h nmr,
13
c nmr,确认其化学结构。(1h nmr(400mhz,cdcl3,25℃):δ7.82(br s,nh),6.57(d,1h,c4h3nh),6.12(m,1h,c4h3nh),6.04(s,1h,c4h3nh),3.95(s,2h,-ch
2-),3.32(s,ch2,c9h7),6.20(s,1h,c9h7),7.42(d,1h,c9h7),7.17-7.26(m,3h,c9h7);
13
c nmr(100mhz,cdcl3):δ143.72,143.41,140.92,128.96,127.85,125.14,123.74,122.72,118.26,115.62,107.27,105.24,36.63,25.65.)
[0075]
(3)络合物1[(η
5-c9h6)ch2(α-c4h3n)]ti(nme2)2的合成
[0076][0077]
取一个100ml schlenk瓶,用加热枪抽烤并充换氩气三次,加入30ml新蒸甲苯和1.95g配体(c9h7)ch2(α-c4h3nh)(10.0mmol),在冰水浴下加入2.25g ti(nme2)4(10mmol),撤去冰水浴,加热到70℃反应4h,整个反应过程中一直在通入少量保护气下进行,保证气体副产物不断被带走,反应结束之后用抽干溶剂,得到黄色微粘稠固体,在正己烷/甲苯=1:1混合溶剂下低温重结晶,得到2.15黄色固体,收率为73%。
[0078]
通过1h nmr,
13
c nmr,元素分析确认其化学结构。(1h nmr(400mhz,cdcl3,25℃):7.45(m,2h,benzo),6.70(m,2h,benzo),6.70(m,1h,c4h3n),5.99(m,1h,c4h3n),5.91(m,1h,c4h3n),6.49(m,2h,c5h2),4.30-4.26(m,1h,ch2),3.98-3.94(m,1h,ch2),3.35(s,6h,(nme2)2,2.24(s,6h,(nme2)2);
13
c nmr(100mhz,cdcl3):δ153.22,126.07,124.58,124.09,124.04,123.57,123.29,121.77,121.49,118.07,105.95,101.24,98.56,49.05,45.17,25.98;anal.found:c,65.65;h,6.99;n,12.77.)
[0079]
实施例2
[0080]
络合物1催化乙烯均相聚合在反应之前,对反应釜进行真空预热,设定温度为80℃后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中加料:催化剂加入量为10μmol,甲苯100ml,mao为6.7ml(即铝钛比为1000);从加料器中加料后,设定乙烯压力0.6mpa,反应30min后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(100ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,得到11.5g固体,计算催化剂活性为3.6
×
106g/(mol
·
ti
·
h)
[0081]
实施例3
[0082]
络合物1催化乙烯与1-己烯共聚合
[0083]
在反应之前,对反应釜进行真空预热,设定温度为80℃后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中加料:催化剂加入量为10μmol,甲苯100ml,mao为6.7ml(即铝钛比为1000),1-己烯20ml;从加料器中加料后,设定乙烯压力0.6mpa,反应30min后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(100ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,得到9.5g固体,计算催化剂活性为1.8
×
106g/(mol
·
ti
·
h),根据聚合物
13
c-nmr谱图计算的到共聚物中1-己烯插入率8.40%。
[0084]
实施例4
[0085]
络合物1催化乙烯与1-辛烯共聚合
[0086]
在反应之前,对反应釜进行真空预热,设定温度为80℃后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中加料:催化剂加入量为10μmol,甲苯100ml,mao为6.7ml(即
铝钛比为1000),1-辛烯23ml;从加料器中加料后,设定乙烯压力0.6mpa,反应30min后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(100ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,得到6.0g固体,计算催化剂活性为1.1
×
106g/(mol
·
ti
·
h),根据聚合物
13
c-nmr谱图计算的到共聚物中1-辛烯插入率7.06%。
[0087]
实施例5
[0088]
络合物2[(η
5-c9h6)ch2(α-c4h3n)]ti(net2)2的合成
[0089]
取一个100ml schlenk瓶,用加热枪抽烤并充换氩气三次,加入40ml新蒸甲苯和1.95g配体(c9h7)ch2(2-c4h3nh)(10.0mmol),在冰水浴下加入3.36g ti(net2)4(10mmol),撤去冰水浴,加热到80℃反应4h,整个反应过程中一直在通入少量保护气下进行,保证气体副产物不断被带走,反应结束之后用抽干溶剂,得到紫红色微粘稠固体,在正己烷/甲苯=1:1混合溶剂下低温重结晶,得到2.24g深红色固体,收率为58%。
[0090]
通过1h nmr,
13
c nmr,元素分析确认其化学结构。(1h nmr(400mhz,cdcl3,25℃):7.34(m,1h,c4h3n),6.04(m,1h,c4h3n),5.96(m,1h,c4h3n),7.15(m,4h,benzo),6.50(m,1h,c5h2),6.13(m,1h,c5h2),4.23(s,2h,ch2),3.52(m,4h,(net2)
2-ch2),3.38(m,4h,(net2)
2-ch2),1.09(m,12h,(net2)
2-ch3);
13
c nmr(100mhz,cdcl3):δ143.73,143.42,140.92,128.98,127.85,125.14,123.74,122.73,118.28,115.63,107.27,105.24,42.44,36.64,25.68;anal.found:c,61.68;h,7.24;n,9.81.)
[0091]
实施例6
[0092]
络合物2催化乙烯均相聚合
[0093]
在反应之前,对反应釜进行真空预热,设定温度为80℃后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中加料:催化剂加入量为10μmol,甲苯100ml,mao为6.7ml(即铝锆比为1000);从加料器中加料后,设定乙烯压力0.6mpa,反应30min后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(100ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,得到22.5g固体,计算催化剂活性为3.6
×
106g/(mol
·
ti
·
h)。
[0094]
实施例7
[0095]
络合物2催化乙烯与1-己烯共聚合
[0096]
在反应之前,对反应釜进行真空预热,设定温度为80℃后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中加料:催化剂加入量为10μmol,甲苯100ml,mao为6.7ml(即铝锆比为1000),1-己烯20ml;从加料器中加料后,设定乙烯压力0.6mpa,反应30min后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(100ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,得到11.5g固体,计算催化剂活性为1.7
×
106g/(mol
·
zr
·
h),根据聚合物
13
c-nmr谱图计算的到共聚物中1-己烯插入率9.81%。
[0097]
实施例8
[0098]
络合物2催化乙烯与1-辛烯共聚合
[0099]
在反应之前,对反应釜进行真空预热,设定温度为80℃后打开氩气将反应釜冲洗三次,然后保持恒温,去手套箱中加料:催化剂加入量为10μmol,甲苯100ml,mao为6.7ml(即铝锆比为1000),1-辛烯23ml;从加料器中加料后,设定乙烯压力0.6mpa,反应30min后,关闭乙烯进气阀门,打开冷凝水降温,打开反应釜取出内衬将产物倒入烧杯中,加入盐酸-乙醇溶液(v盐酸:v乙醇=1:15)终止反应。将聚合物先用盐酸-乙醇溶液反复冲洗,溶解掉反应残留下来的铝盐,接着用去离子水再清洗三次(100ml
×
3),最后放入真空干燥箱,60℃下干燥至恒重,得到6.5g固体,计算催化剂活性为1.2
×
106g/(mol
·
ti
·
h),根据聚合物
13
c-nmr谱图计算的到共聚物中1-辛烯插入率8.84%。
[0100]
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1