3-噻唑烯基氟化硼络合二吡咯甲川类化合物及其制备方法和用途与流程
本发明涉及荧光探针,具体涉及一类3-噻唑烯基氟化硼络合二吡咯甲川(bodipy)类化合物、其制备方法及其用于制备荧光探针的用途。
背景技术:
阿尔茨海默症(alzheimer’sdisease,ad)是一种进行性发展的神经退行性疾病。在病理学中,淀粉样蛋白肽(β-amyloid,aβ)聚集后沉积在大脑边缘和大脑皮层形成的老年斑(senileplaques,sp)是ad最典型的病理特征之一。特异性识别aβ蛋白的探针是诊断ad的重要工具,2012-2014年fda批准上市了三个靶向识别aβ蛋白的放射性同位素18f标记探针florbetapir、flutemetamol和florbetaben,作为造影剂用于正电子发射断层扫描成像(positronemissiontomography,pet)诊断ad。但是pet诊断价格昂贵,需要临时制备放射性同位素标记的诊断试剂(11c半衰期为20min,18f半衰期为110min)。与pet相比,荧光成像技术具有灵敏度高,快速实时成像,成本低和无放射性的特点。
近年来,aβ成像的荧光探针研究逐渐增多,已报道的aβ识别的荧光探针(代表性的结构如下)具有结构多样性,其中一些探针被证明可以有效用于ad的活体诊断。但该类探针的量子产率均较低(一般不超过10%),部分染料的血脑屏障透过性差,生物相容性不好。
化学结构如下的氟化硼络合二吡咯甲川(bodipy)荧光染料具有化学稳定性高、光稳定性高和生物相容性好的优势,在生物及医学领域具有广泛的应用。
因此,有必要从bodipy中开发一种量子产率高、生物相容性优良的荧光探针。
技术实现要素:
本发明的一个目的是提供一类3-位为取代的噻唑烯基的新结构化合物,其具有量子产率高、光学稳定性好和生物相容性优异的特点。
本发明的另一个目的是提供上述化合物的制备方法。
本发明的再一个目的是提供上述化合物用于识别aβ蛋白的用途。
根据本发明的目的,本发明提供了一类如通式ⅰ所示的氟化硼络合二吡咯甲川(bodipy)类化合物:
其中,r1、r2各自独立选自氢、c1-c6直链或支链烷基,或r1和r2与其相连的氮原子连接形成5-7元含氮杂环,可选地,所述5-7元含氮杂环可进一步含有除氮以外的杂原子,例如还含有氧杂原子;
r3、r4、r5、r6、r7各自独立选自氢、c1-c6直链或支链烷基,
r8选自氢、c1-c6直链或支链烷基、r9-c1-c6直链或支链烷基、5-7元芳香基团或c3-c10环烷基,
r9选自卤素、r10c(=o)o-、r11r12n-、r13c(=o)n-、r14c(=o)-、r15-n-c(=o)或r16o-,
r10和r13各自独立选自c1-c6直链或支链烷基、卤素取代的c1-c6直链或支链烷基、5-7元芳香基团,
r11、r12和r15各自独立选自氢、c1-c6直链或支链烷基、卤素取代的c1-c6直链或支链烷基、5-7元芳香基团;
r14选自氢、羟基、c1-c6直链或支链烷基、卤素取代的c1-c6直链或支链烷基、c1-c6直链或支链烷氧基、卤素取代的c1-c6直链或支链烷氧基、5-7元芳香基团,
r16选自氢、c1-c6直链或支链烷基、用卤素或c1-c6直链或支链烷氧基取代的c1-c6直链或支链烷基、5-7元芳香基团,
“-”为负电荷,“+”为正电荷。
在本申请中,
c1-c6直链或支链烷基包括具有1、2、3、4、5或6个碳原子的直链或支链烷基,非限制性地包括甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、正戊基、新戊基、异戊基、己基等,优选为c1-c4直链或支链烷基。
c1-c6直链或支链烷氧基包括具有1、2、3、4、5或6个碳原子的直链或支链烷氧基,非限制性地包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、叔丁氧基、异丁氧基、正戊氧基、新戊氧基、异戊氧基、己氧基等,优选为c1-c4直链或支链烷氧基。
5-7元含氮杂环指的是环上包括至少一个氮原子且具有5-7个环原子的杂环,非限制性地包括吡咯烷环、哌啶环、吗啉环、哌嗪环、高哌啶环、氮氧杂环戊烷等。
5-7元芳香基团指的是环上具有5-7个环原子且具有芳香性的环状基团,非限制性地包括苯基、吡啶基、噻吩基、噻喃基、噻唑基、吡喃基、吡咯基等。
c3-c10环烷基指的是环上具有3-10个环碳原子的饱和环状烷基,非限制性地包括环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基、环癸基等。
所述卤素可为f、cl、br或i。
在具体实施方式中,r1或r2各自独立地选自氢、甲基、乙基,或者r1和r2与其相连的氮原子连接形成
在具体实施方式中,r3、r4、r5、r6、r7各自独立选自氢、甲基或乙基。在一个实施方式中,r5、r6、r7都是甲基。
在具体实施方式中,r8为氢、甲基、环丙基、丙基、丁基、苯基、
在实施方式中,所述bodipy类化合物可以选自以下各化合物:
另一方面,本发明提供了上述通式i所示的bodipy类化合物的制备方法,所述方法包括以下步骤:
(a)原料1与胺化合物2在碱性条件下发生亲核取代得到中间体3;
(b)当吡咯化合物4与吡咯化合物5相同时,吡咯化合物4与r8cocl发生friedel–crafts反应,再向反应溶液中加入三氟化硼-乙醚复合物和碱,得到中间体6;
当吡咯化合物4与吡咯化合物5不同时,1eq.的吡咯化合物4或5首先与r8cocl发生friedel–crafts反应,反应完成后旋干溶剂,再与1eq.的吡咯化合物5或4在酸或三氯氧磷催化条件下发生缩合反应,最后向反应溶液中加入三氟化硼-乙醚复合物和碱,得到中间体6;
(c)中间体6与中间体3在乙酸和哌啶催化条件下发生缩合反应得到通式i的化合物,
其中,r1至r8的定义如前所述。
具体地,在上述步骤(a)中,原料1溶于水和dmso混合溶液,依次加入胺2和碱反应。优选地,该反应可在加热条件下进行,例如,加热至50-100℃,使用的碱可选自三乙胺、二异丙基乙胺、碳酸钠或碳酸钾,优选碳酸钾。
具体地,在上述步骤(b)中,当吡咯化合物4与吡咯化合物5不同时,吡咯化合物4或5溶于无水二氯甲烷,碱性条件下缓慢加入酰氯r8cocl发生friedel–crafts反应,旋干反应溶剂,产物进一步溶于氯代烷烃,如二氯甲烷、氯仿或二氯乙烷,冰浴条件下加入三氟乙酸、三氟甲磺酸或三氯氧磷,升至室温反应。反应完成后,再向反应液中加入三氟化硼-乙醚复合物和碱。所述加入三氟化硼-乙醚复合物的反应可在加热条件下进行,例如,加热至50-70℃,使用的碱可选自三乙胺、二异丙基乙胺、吡啶或碳酸钾,优选三乙胺或二异丙基乙胺。
具体地,在上述步骤(c)中,中间体6与中间体3溶于无水甲苯,加入少量氯仿或二氯甲烷助溶,再加入催化量的乙酸和哌啶,加热回流,油水分离器分水反应,得到通式i所示的化合物。
再一方面,本发明提供了上述通式ⅰ所示的bodipy类化合物在制备荧光探针试剂中的用途,特别地,所述荧光探针试剂可用于识别aβ蛋白。
在具体实施方式中,所述bodipy类化合物可被用作荧光探针用于体外和体内识别aβ蛋白。
在具体实施方式中,所述试剂为用于诊断阿尔茨海默病或监控阿尔茨海默病疗效的试剂。
再一方面,本发明提供了一种识别aβ蛋白的方法,所述方法包括,使用上述通式ⅰ所示的bodipy类化合物作为荧光探针来识别aβ蛋白。
再一方面,本发明提供了一种诊断阿尔茨海默病或监控阿尔茨海默病疗效的方法,所述方法包括,使用上述通式ⅰ所示的bodipy类化合物作为荧光探针来识别aβ蛋白,以诊断阿尔茨海默病或监控阿尔茨海默病疗效。
再一方面,本发明提供一种药物组合物,其包含上述bodipy类化合物。所述组合物可以用作诊断阿尔茨海默病或监控阿尔茨海默病疗效的试剂。
在具体实施方式中,上述药物组合物可配制为口服剂型。
有益效果
本发明通过在bodipy类化合物的3-位引入烯基噻唑结构,提供了一类具有新结构的化合物,其具有量子产率高(乙醇中量子产率>25%)、光学稳定性好的特点。此外,该类化合物作为荧光探针对aβ蛋白具有高亲和力,具有体外识别aβ的能力,而且由于其可快速通过血脑屏障,在体内也具有识别aβ的能力,因此其生物相容性好,可以通过口服用于阿尔茨海默病的体内诊断和疗效监控。
附图说明
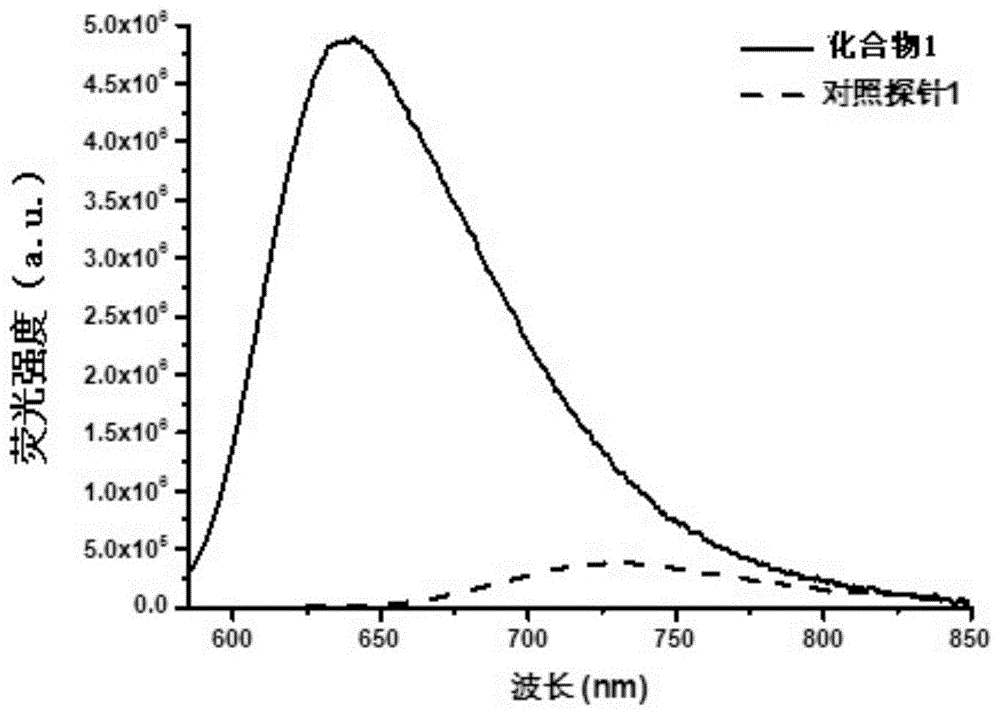
图1:制备例1合成的化合物1和对照探针1在乙醇中的荧光发射光谱(5μm)。
图2:制备例1合成的化合物1对aβ1-42纤维kd值测试的曲线图。
图3:制备例1合成的化合物1特异性标记app/ps1转基因鼠脑片上的老年斑(化合物1的激发光584nm,发射光641nm;硫磺素s(ths)的激发光488nm,发射光550nm;比例尺250μm)。
图4:a:制备例1合成的化合物1在小鼠中的成像结果(其中roi7=1.501x107,roi8=5.472x106),b:制备例1合成的化合物1在小鼠脑部不同时间点的平均发光强度。
具体实施方式
下面的实施例用于具体地说明本发明化合物,其制备方法,以及其作为开关型荧光探针分子的应用,但本发明并不局限于这些实施例。
核磁共振氢谱(1hnmr)用brukeramx-400型、gemini-300型或amx–600型核磁共振仪记录,溶剂为氘代氯仿(cdcl3)。化学位移的单位为ppm,耦合常数j的单位为hz。所有反应溶剂均按照常规方法进行纯化。柱层析用硅胶(200-300目)为青岛海洋化工分厂生产。薄层层析使用gf254高效板,为烟台化工研究所生产。制备型薄层层析板自制,固定相采用gf254(hg/t2354-92)硅胶和羧甲基纤维素钠(800-1200)制备,分别为青岛海洋化工有限公司和中国医药(集团)上海化学试剂公司生产。所有溶剂均为分析纯试剂,所用试剂均购自国药集团化学试剂有限公司。采用紫外荧光等方法显色。减压蒸除有机溶剂在旋转蒸发仪中进行。
制备实施例
通式i化合物的通用合成方法
步骤1:
2-溴噻唑-5-甲醛1(1eq.)溶于h2o:dmso=5:1的混合溶剂中,依次加入胺2(2eq.)和碳酸钾(3eq.)(若胺为盐,加入5eq的碳酸钾),氮气保护下加热至50℃反应3-6h。反应液冷却,倒入冰水中,二氯甲烷萃取,硫酸钠干燥,旋干溶剂,过柱纯化,获得产品,白色至浅棕色固体,产率60-90%。
步骤2:
当吡咯化合物4与吡咯化合物5不同时,反应如下:
吡咯化合物4或5(1eq)溶于无水二氯甲烷,冰浴条件下缓慢加入酰氯r8cocl(1eq),升至室温反应0.5-4h,tlc检测。反应完成后旋干溶剂,复溶于无水氯仿,氮气保护,冰浴条件下依次加入取代吡咯化合物5或4(1eq.)和三氯氧磷(1eq.),升至室温反应3-6h。tlc监测反应完成后冰浴冷却反应液,依次缓慢加入三乙胺(10eq.)和三氟化硼乙醚(10eq.)。50℃加热反应1-8h,反应液冷却,倒入冰水中淬灭,二氯甲烷萃取,硫酸钠干燥,旋干溶剂,过柱纯化,获得产品,红棕色固体,产率10-40%。
当上述吡咯化合物4与吡咯化合物5相同时,反应如下:
圆底烧瓶中加入取代吡咯化合物4(2.2eq.),溶于无水二氯甲烷,氮气保护,冰浴,用恒压低液漏斗滴入酰氯r8cocl(7)(1.0eq.),滴加完成后加热回流反应8h,旋干二氯甲烷,加入甲苯和二氯甲烷(甲苯与二氯甲烷体积比=19:1),冰浴,加入三乙胺(7.0eq.),用恒压低液漏斗滴入三氟化硼-乙醚络合物(7.0eq.),滴加完成后,去除冰浴,室温反应10-20min,50℃加热反应3h,反应液冷却,倒入冰水中淬灭,二氯甲烷萃取,硫酸钠干燥,旋干溶剂,过柱纯化,获得产品,红棕色固体,产率20-40%。
步骤3:
中间体6(1eq)溶于无水甲苯,加入少量二氯甲烷或氯仿助溶,依次加入取代的噻唑甲醛3(1eq)、哌啶(0.2eq)和乙酸(0.2eq),氮气保护,分水器装置,回流反应2h。旋干溶剂,柱层析分离,得深色固体(20-50%)。
制备例1:化合物1的合成
步骤1:
将5-溴噻唑-1-甲醛(2.0g,10.47mmol)溶于20ml二甲胺的水溶液(33%w/w),回流反应4h。加入50ml乙酸乙酯萃取,有机相依次用水洗、饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋干溶剂,柱层析分离(pe:ea=10:1),得到产品,浅色固体(1.3g,81%)。1hnmr(300mhz,cdcl3)δ9.48(s,1h),7.47(d,j=4.4hz,1h),5.92(d,j=4.4hz,1h),3.08(s,6h).
步骤2:
将2,4-二甲基吡咯(7.0g,73.6mmol)溶于200ml无水二氯甲烷(dcm)中,冰浴条件下滴入4-溴丁酰氯(6.75g,36.4mmol),回流反应5h。旋干溶剂,残渣用200ml甲苯(toluene)/dcm=19:1的混合溶剂溶解,依次加入三乙胺(tea,18.4g,182mmol)和三氟化硼乙醚复合物(bf3·et2o,36.2g,255mmol),50℃反应1h,将反应液倒入400ml冰水,加入200ml二氯甲烷萃取,有机相水洗两次,无水硫酸钠干燥,旋干溶剂,柱层析分离,得到产物(4.0g,36%)。1hnmr(400mhz,cdcl3)δ6.06(s,2h),3.78(t,j=5.9hz,2h),3.05-2.96(m,2h),2.53(s,6h),2.41(s,6h),1.89(s,1h),1.85-1.78(m,2h);
步骤3:
将步骤2产物(500mg,1.63mmol)溶于70ml二氯甲烷/甲苯(toluene)=1:6的混合溶剂中,加入步骤1产物(253mg,1.63mmol)、哌啶(piperdine,100ul)和乙酸(acoh,100ul),氮气保护,分水器装置,回流反应2h。旋干溶剂,柱层析分离(pe:ea:dcm=5:1:3),蓝紫色固体(246mg,35%)。1hnmr(400mhz,cdcl3)δ7.52(s,1h),6.90(d,j=3.9hz,1h),6.57(s,1h),6.00(s,1h),5.77(d,j=4.0hz,1h),3.77(s,2h),3.29-3.22(m,1h),3.01(s,6h),2.58-2.47(m,5h),2.42(s,3h),2.39(s,3h),1.83(s,2h).
制备例2:化合物2的合成
将化合物1溶于无水二氯甲烷,依次加入乙酸酐和三乙胺,室温反应1h。加水淬灭反应,二氯甲烷萃取,无水硫酸钠干燥,旋干溶剂,柱层析分离(pe:ea:dcm=10:1:3),蓝紫色固体(47%)。1hnmr(400mhz,cdcl3)δ7.55(s,1h),6.93(d,j=4.1hz,1h),6.29(s,1h),6.10(s,1h),5.47(d,j=4.0hz,1h),3.89(s,2h),3.21(s,6h),2.74-2.52(m,5h),2.40(s,3h),2.29(s,3h),2.04(s,3h),1.85(s,2h).
制备例3:化合物3的合成
将化合物1溶于无水二氯甲烷,依次加入4-溴丁酰氯和三乙胺,室温反应1h。加水淬灭反应,二氯甲烷萃取,无水硫酸钠干燥,旋干溶剂,柱层析分离(pe:ea:dcm=10:1:3),蓝紫色固体(53%)。1hnmr(400mhz,cdcl3)δ7.51(s,1h),6.70(d,j=4.1hz,1h),6.47(s,1h),6.03(s,1h),5.45(d,j=4.0hz,1h),4.13(t,j=13.6hz,2h),3.55(m,2h),3.21(s,6h),2.62(s,3h),2.56–2.47(m,7h),2.25(s,3h),2.13–1.93(m,2h),1.71(s,2h).
制备例4:化合物4的合成
化合物2溶于无水甲醇,加入胺的甲醇溶液,室温反应8h。旋干溶剂,柱层析分离,得蓝紫色固体(17%)。1hnmr(400mhz,cdcl3)δ7.42(s,1h),6.70(d,j=3.9hz,1h),6.47(s,1h),6.07(s,1h),5.87(d,j=4.0hz,1h),3.11(s,6h),2.66(t,j=10.3hz,2h),2.58-2.47(m,5h),2.35(s,3h),2.41-2.23(m,5h),1.93(s,2h).
制备例5:化合物5的合成
合成方法同化合物2,不同之处在于用化合物4替换化合物1作为起始反应物,产物为蓝紫色固体,产率45%。1hnmr(400mhz,cdcl3)δ7.43(s,1h),7.31(t,j=5.4hz,1h),6.91(d,j=3.9hz,1h),6.67(s,1h),6.00(s,1h),5.77(d,j=4.0hz,1h),3.11(d,j=5.6hz,2h),3.01(s,6h),2.66(t,j=10.3hz,2h),2.58(s,3h),2.45(s,3h),2.41-2.23(m,5h),1.89(s,3h).
制备例6:化合物6的合成
合成方法同化合物1,不同之处在于将化合物1合成步骤1中的二甲胺替换成甲基乙基胺。1hnmr(400mhz,cdcl3)δ7.72(s,1h),6.91(d,j=3.9hz,1h),6.57(s,1h),6.20(s,1h),5.77(d,j=4.0hz,1h),3.77(s,2h),3.29-3.22(m,1h),3.03(q,j=12.6hz,2h),2.98(s,3h),22.58-2.47(m,5h),2.41(s,3h),2.39(s,3h),1.83(s,2h),1.10(t,j=12.5hz,3h).
制备例7:化合物7的合成
合成方法同化合物1,不同之处在于将化合物1合成步骤1中的二甲胺替换成二乙胺。1hnmr(400mhz,cdcl3)δ7.49(s,1h),6.89(d,j=3.9hz,1h),6.47(s,1h),5.98(s,1h),5.77(d,j=4.0hz,1h),3.47(s,2h),3.29-3.22(m,1h),3.12(q,j=12.6hz,4h),2.58-2.47(m,5h),2.32(s,3h),2.29(s,3h),1.83(s,2h),1.10(t,j=12.5hz,6h).
制备例8:化合物8的合成
合成方法同化合物1,不同之处在于将化合物1合成步骤1中的二甲胺替换成吗啉。1hnmr(400mhz,cdcl3)δ7.53(s,1h),6.93(d,j=3.9hz,1h),6.47(s,1h),6.05(s,1h),5.77(d,j=4.0hz,1h),3.87(s,2h),3.68(t,j=9.4hz,4h),3.29-3.22(m,1h),3.20(t,j=9.3hz,4h),2.58-2.47(m,5h),2.43(s,3h),2.39(s,3h),1.83(s,2h).
制备例9:化合物9的合成
合成方法同化合物1,不同之处在于将化合物1合成步骤1中的二甲胺替换成哌啶。1hnmr(400mhz,cdcl3)δ7.62(s,1h),6.91(d,j=3.9hz,1h),6.57(s,1h),6.02(s,1h),5.77(d,j=4.0hz,1h),3.97(s,2h),3.29-3.22(m,1h),3.16(t,j=5.3hz,4h),2.58-2.47(m,5h),2.42(s,3h),2.39(s,3h),1.83(s,2h),1.71–1.49(m,6h).
制备例10:化合物10的合成
合成方法同化合物1,不同之处在于将化合物1合成步骤1中的二甲胺替换成四氢吡咯。1hnmr(400mhz,cdcl3)δ7.55(s,1h),6.90(d,j=3.9hz,1h),6.50(s,1h),6.00(s,1h),5.74(d,j=4.0hz,1h),3.73(s,2h),3.29-3.22(m,1h),3.16–3.02(m,4h),2.58-2.47(m,5h),2.42(s,3h),2.39(s,3h),2.02–1.85(m,4h),1.83(s,2h).
制备例11:化合物11的合成
圆底烧瓶中加入实施例1得到的产品(1.0eq.),溶于无水二氯甲烷,氮气保护,冰浴,加入dmp(1.0eq.),撤出冰浴,室温反应30-60min,加碳酸钠水淬灭反应,二氯甲烷萃取,硫酸钠干燥,旋干溶剂,过柱纯化,获得产品,蓝紫色固体,产率36%,1hnmr(400mhz,cdcl3)δ9.85(t,j=14.0hz,1h),7.42(s,1h),6.87(d,j=3.9hz,1h),6.57(s,1h),5.99(s,1h),5.77(d,j=4.0hz,1h),3.04(s,6h),2.47(s,3h),2.42(s,3h),2.39-2.23(m,5h),1.85(t,j=11.0hz,2h).
制备例12:化合物12的合成
步骤1:
将丁二酸酐(10.0g,105mmol)溶于300ml无水二氯甲烷(dcm)中,氮气保护下,加入2,4-二甲基吡咯(6.31g,63mmol),冰浴条件下加入三氟化硼乙醚络合物(bf3·et2o,17.9g,126mmol),回流反应8h。反应液在冰浴条件下依次加入三乙胺(tea,46.1g,420mmol)和三氟化硼乙醚络合物(bf3·et2o,44.8g,315mmol),50℃反应2h。将反应液缓慢倒入400ml冰水中,加入300ml二氯甲烷萃取,有机相用水洗两次,无水硫酸钠干燥,旋干溶剂,柱层析纯化(pe:ea:dcm=10:2:5),得到产物为深红色固体(2.5g,16%),1hnmr(cdcl3,400mhz)δ6.08(s,2h),3.34(t,j=8.8hz,2h),2.67(t,j=8.8hz,2h),2.85(s,6h),2.53(s,6h)。
步骤2:
合成方法同化合物1合成的步骤3,不同之处在于将化合物1合成的步骤3的中间体6替换成上述步骤1的产物。1hnmr(400mhz,dmso)δ10.56(t,j=14.0hz,1h),7.52(s,1h),6.90(d,j=3.9hz,1h),6.57(s,1h),6.00(s,1h),5.77(d,j=4.0hz,1h),3.04(s,6h),2.47(s,3h),2.42(s,3h),2.40-2.24(m,5h),1.86(t,j=11.1hz,2h).
制备例13:化合物13的合成
将化合物12溶于无水二氯己烷,依次加入edci、hoat、三乙胺和甲胺盐酸盐,室温反应2h。加水淬灭反应,二氯甲烷萃取,无水硫酸钠干燥,旋干溶剂,柱层析纯化,得到产物,紫黑色固体(16%),1hnmr(400mhz,cdcl3)δ7.62(s,1h),7.03(s,1h),6.93(d,j=3.9hz,1h),6.47(s,1h),6.00(s,1h),5.76(d,j=4.0hz,1h),3.04(s,6h),2.80(s,2h),2.37(s,3h),2.32(s,3h),2.40-2.24(m,5h),1.83(t,j=11.3hz,2h).
制备例14:化合物14的合成
步骤1:
除了以2,4-二甲基吡咯(5.0g,52.6mmol)和5-溴戊酰氯(5.2g,26.3mmol)为原料以外,具体操作同化合物1的合成步骤2,深棕色固体(4.6g,46%)。1hnmr(400mhz,cdcl3)δ6.09(s,2h),3.61(t,j=6.3hz,0.5h),3.47(t,j=6.5hz,1.5h),3.03–2.96(m,2h),2.54(s,6h),2.45(s,6h),2.13–2.03(m,1.5h),1.98(dd,j=14.1,6.7hz,0.5h),1.83(m,2h).
步骤2:
合成方法同化合物1的合成步骤3。不同之处在于将化合物1的合成步骤3中的化合物6替换成上述步骤1中的产物。1hnmr(400mhz,cdcl3)δ7.51(s,1h),6.91(d,j=3.9hz,1h),6.54(s,1h),6.01(s,1h),5.77(d,j=4.0hz,1h),3.47(t,j=7.1hz,2h),3.03(s,6h),2.53(s,3h),2.42(s,3h),2.35-2.40(m,5h),1.94(t,j=7.1hz,2h),1.83(m,2h).
制备例15:化合物15的合成
将化合物14(1eq)溶于无水dmf,加入无水乙酸钠(2eq),50℃反应8h。加入二氯甲烷稀释反应液,有机相水洗两次,无水硫酸钠干燥,旋干溶剂,柱层析分离,得到产物,蓝紫色固体(23%)。1hnmr(400mhz,cdcl3)δ7.55(s,1h),6.96(d,j=3.9hz,1h),6.57(s,1h),6.10(s,1h),5.77(d,j=4.0hz,1h),4.12(s,2h),3.04(s,6h),2.58-2.47(m,5h),2.42(s,3h),2.39(s,3h),2.10(s,3h),1.75-1.53(m,4h).
制备例16:化合物16的合成
将化合物15(1eq)溶于四氢呋喃和甲醇混合溶剂(4:1),加入氢氧化锂(1.2eq)反应1h。加入二氯甲烷稀释反应液,有机相水洗两次,无水硫酸钠干燥,旋干溶剂,柱层析分离,得到产物,蓝紫色固体(37%)。1hnmr(400mhz,cdcl3)δ7.52(s,1h),6.92(d,j=3.9hz,1h),6.56(s,1h),6.10(s,1h),5.77(d,j=4.0hz,1h),3.78(s,2h),3.29-3.22(m,1h),3.04(s,6h),2.58-2.47(m,5h),2.43(s,3h),2.39(s,3h),1.70-1.52(m,4h).
制备例17:化合物17的合成
合成方法同化合物1。不同之处在于将化合物1合成步骤2的原料2,4-二甲基吡咯替换成2,4-二甲基-3-乙基吡咯,得紫黑色固体,总产率8%。1hnmr(400mhz,cdcl3)δ7.29(s,1h),6.91(d,j=3.9hz,1h),5.78(d,j=4.0hz,1h),3.77(m,2h),3.25(s,1h),3.01(s,6h),2.61-2.47(m,7h),2.45-2.29(m,5h),2.39(s,3h),1.83(m,2h),1.18(t,j=8.0hz,3h),1.07(t,j=8.0hz,3h)
制备例18:化合物18的合成
合成方法同化合物1,不同之处在于吡咯化合物4为2,4-二甲基吡咯,吡咯化合物5为吡咯,蓝紫色固体,总产率15%。1hnmr(400mhz,cdcl3)δ7.60(s,1h),7.52(s,1h),6.91(d,j=3.9hz,1h),7.07(d,j=4.0hz,1h),6.42(dd,j=4.1,2.1hz,1h),6.14(s,1h),5.78(d,j=4.0hz,1h),3.78(t,j=8.0hz,2h),3.01(s,6h),2.97-2.85,(m,2h),2.45(s,3h),1.89(s,1h),1.85-1.78,(m,2h),
制备例19:化合物19的合成
合成步骤同化合物18,不同之处在于将2,4-二甲基吡咯替换成2,4-二甲基-3-乙基吡咯,紫黑色固体,总产率6%。1hnmr(500mhz,cdcl3)δ7.29(s,1h),7.14(d,j=10.2hz,1h),6.61(d,j=10.2hz,1h),5.62(m,1h),4.43(dd,j=21.8,12.3hz,1h),4.14(dd,j=21.8,1.8hz,1h),3.53(t,j=15.0hz,2h),3.13(s,6h),2.59(q,j=13.2hz,2h),2.38(t,j=15.6hz,2h),2.13(s,3h),1.79–1.53(m,2h),1.20(dd,j=37.0,23.8hz,4h).
制备例20:化合物20的合成
合成步骤同化合物18,不同之处在于将吡咯替换成2,4-二甲基-3-乙基吡咯,蓝紫色固体,总产率9%。1hnmr(400mhz,cdcl3)δ7.53(s,1h),6.91(d,j=3.9hz,1h),6.03(s,1h),5.77(d,j=4.0hz,1h),3.76(t,j=8.0hz,2h),3.03(s,6h),2.97-2.85,(m,2h),2.53(s,3h),2.42(s,3h),2.35-2.40(m,5h),1.89(s,1h),1.85-1.78,(m,2h),1.06(t,j=8.0hz,3h).
制备例21:化合物21的合成
合成步骤同化合物1,不同之处在于将4-溴丁酰氯替换成4-甲氧基丁酰氯,蓝紫色固体,总产率13%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),6.92(d,j=3.9hz,1h),6.54(s,1h),6.03(s,1h),5.77(d,j=4.0hz,1h),3.76(t,j=8.0hz,2h),3.62(s,3h),3.02(s,6h),2.97-2.85,(m,2h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.85-1.76,(m,2h)
制备例22:化合物22的合成
合成方法同化合物2,不同之处在于将乙酸酐替换为苯基酸酐,蓝紫色固体,产率46%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),δ7.28(dd,j=7.4,1.5hz,2h),7.10–7.06(m,1h),6.97(t,j=7.5hz,2h),6.92(d,j=3.9hz,1h),6.54(s,1h),6.03(s,1h),5.77(d,j=4.0hz,1h),3.66(t,j=8.0hz,2h),3.02(s,6h),2.97-2.85,(m,2h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.85-1.76,(m,2h)
制备例23:化合物23的合成
合成方法同化合物1,不同之处在于将4-溴丁酰氯替换成4-苯氧基丁酰氯,蓝紫色固体,总产率11%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),δ7.33(dd,j=7.4,1.5hz,2h),7.00(t,j=7.4hz,1h),6.97(t,j=7.5hz,2h),6.92(d,j=3.9hz,1h),6.54(s,1h),6.06(s,1h),5.77(d,j=4.0hz,1h),4.12(t,j=8.0hz,2h),3.02(s,6h),2.97-2.85,(m,2h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.85-1.76,(m,2h)
制备例24:化合物24的合成
苯胺(1.3eq.)溶于1,2-二氯乙烷/甲醇的混合溶液中,依次加入化合物11(1.0eq.)、氰基硼氢化钠(1.1eq.)和1滴冰乙酸,室温反应6h。旋干溶剂,柱层析分离,蓝黑色固体,产率33%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),δ7.17(dd,j=7.4,1.5hz,2h),6.92(d,j=3.9hz,1h),6.71(t,j=7.3hz,1h),6.59(t,j=7.5hz,2h),6.54(s,1h),6.06(s,1h),5.77(d,j=4.0hz,1h),3.71(s,1h),3.30(t,j=8.0hz,2h),3.02(s,6h),2.97-2.85,(m,2h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.85-1.76,(m,2h)
制备例25:化合物25的合成
合成方法同化合物13,不同之处在于将甲胺替换为苯胺,蓝紫色固体,产率23%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),δ7.14(dd,j=7.4,1.5hz,2h),6.92(d,j=3.9hz,1h),6.81(t,j=7.3hz,1h),6.69(t,j=7.5hz,2h),6.54(s,1h),6.06(s,1h),5.77(d,j=4.0hz,1h),3.51(s,1h),3.34(t,j=8.0hz,2h),3.02(s,6h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.85-1.76,(m,2h)
制备例26:化合物26的合成
化合物1(1eq)溶于无水二氯甲烷,依次加入三乙胺(1.5eq)和对甲苯磺酰氯(1.2eq),室温反应2h,旋干溶剂,加入dmf复溶,再加入碳酸钾(2eq),室温反应1h既得产物,蓝紫色固体,30%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),6.92(d,j=3.9hz,1h),6.54(s,1h),6.03(s,1h),5.77(d,j=4.0hz,1h),3.02(s,6h),2.67-2.62,(m,1h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.75-1.69,(m,2h),1.45-1.39,(m,2h)
制备例27:化合物27的合成
合成方法同化合物1,不同之处在于将4-溴丁酰氯替换为4-甲氧乙氧基丁酰氯,蓝紫色固体,总产率8%。1hnmr(400mhz,cdcl3)δ7.53(s,1h),6.92(d,j=3.9hz,1h),6.54(s,1h),6.03(s,1h),5.77(d,j=4.0hz,1h),4.12–4.08(m,4h),3.76(t,j=8.0hz,2h),3.58(s,3h),3.02(s,6h),2.97-2.85,(m,2h),2.53(s,3h),2.42(s,3h),2.38(s,3h)1.85-1.76,(m,2h)
制备例28:化合物28的合成
合成方法同化合物1,不同之处在于将4-溴丁酰氯替换为乙酰氯,蓝紫色固体,总产率6%。1hnmr(400mhz,cdcl3)δ7.50(s,1h),6.92(d,j=3.9hz,1h),6.54(s,1h),6.03(s,1h),5.77(d,j=4.0hz,1h),3.02(s,6h),2.53(s,3h),2.42(s,3h),2.38(s,3h),2.28(s,3h).
制备例29:化合物29的合成
合成方法同化合物1,不同之处在于将4-溴丁酰氯替换为苯甲酰氯,蓝紫色固体,总产率8%。1hnmr(400mhz,cdcl3)δ7.60(s,1h),7.49-7.47(m,3h),7.29-7.26(m,2h),6.82(d,j=3.9hz,1h),6.44(s,1h),6.00(s,1h),5.77(d,j=4.0hz,1h),3.02(s,6h),2.53(s,3h),2.42(s,3h),2.38(s,3h).
制备例30:化合物30的合成
合成方法同实施例2,不同之处在于将吡咯化合物4与4-溴丁酰氯反应的产物替换为商业可购买的3,5-二甲基吡咯-2-甲醛,蓝紫色固体,总产率5%。1hnmr(400mhz,cdcl3)δ7.60(s,1h),7.22(s,1h),6.82(d,j=3.9hz,1h),6.44(s,1h),6.00(s,1h),5.77(d,j=4.0hz,1h),3.02(s,6h),2.53(s,3h),2.42(s,3h),2.38(s,3h).
对照探针1:
对照探针1是bodipy类aβ蛋白识别荧光染料bap-2的类似物。
对照探针1的合成方法如下:
步骤1:
将5-溴噻吩-2-甲醛(2.0g,10.47mmol)溶于20ml二甲胺的水溶液(33%w/w),回流反应4h。加入50ml乙酸乙酯萃取,有机相依次用水洗、饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋干溶剂,柱层析分离(pe:ea=10:1),得到5-二甲基氨基-噻吩-2甲醛,浅色固体(1.3g,81%)。1hnmr(300mhz,cdcl3)δ9.48(s,1h),7.47(d,j=4.4hz,1h),5.92(d,j=4.4hz,1h),3.08(s,6h).
步骤2:
将化合物1的合成步骤2的产物(500mg,1.63mmol)溶于70ml二氯甲烷/甲苯(toluene)=1:6的混合溶剂中,加入5-二甲基氨基-噻吩-2甲醛(253mg,1.63mmol)、哌啶(piperdine,100ul)和乙酸(acoh,100ul),氮气保护,分水器装置,回流反应2h。旋干溶剂,柱层析分离(pe:ea:dcm=10:1:3),紫黑色固体(230mg,32%)。1hnmr(300mhz,cdcl3)δ7.24(d,j=13.4hz,1h),6.99(d,j=15.7hz,1h),6.90(d,j=3.9hz,1h),6.57(s,1h),6.00(s,1h),5.77(d,j=4.0hz,1h),3.77(s,2h),3.29–3.22(m,1h),3.01(s,6h),2.58–2.47(m,5h),2.42(s,3h),2.39(s,3h),1.83(s,2h);
测试实施例
测试例1:代表性化合物1的荧光光谱
将化合物1溶于dmso制成0.5mmol的母液,用乙醇稀释到5μmol,荧光探针的最大激发波长一般为580-650nm,检测波长为600-850nm。代表性化合物1和对照探针1的荧光发射光谱(参见图1)结果显示:化合物1的最大激发波长为590nm,最大发射波长为629nm;对照探针1的最大激发波长为625nm,最大发射波长为715nm。
测试例2:化合物1-30的量子产率
将化合物1-30分别溶于dmso制成0.5mmol的母液,用乙醇稀释到1μmol,测试uv-vis吸收光谱和荧光发射光谱。以乙醇中罗丹明6g的量子产率(φ=0.95,激发波长530nm)为标准测试化合物的相对量子产率,计算公式如下:
其中ф是量子产率,σf为荧光强度积分,abs是吸光度,n为溶剂的折光系数。测试结果如以下表1所示。
表1化合物1-30和对照探针1在乙醇中的量子产率
如表1所示,化合物1-30在乙醇中的荧光强度高,量子产率均大于25%。其中化合物1在乙醇中的量子产率高达52.42%,而对照探针1的量子产率仅为1.83%。化合物1在乙醇中的量子产率是对照探针1的28.6倍,表明噻唑的引入对荧光化合物的量子产率提高具有显著的优势。
测试例3:化合物1-30与aβ1-42纤维亲和力测试
aβ纤维的制备
将aβ1-42(1.0mg,杭州中肽)溶于1ml六氟异丙醇,均分成10份于0.5mlep管中,室温12h挥干有机溶剂,真空干燥2h后,冻存于-20℃冰箱备用。取一支样品,aβ1-42溶于11μldmso和432μl10mmolph=7.4的pbs(aβ1-42终浓度为50μmol)。样品于37℃恒温箱中震荡孵育24h,再加入443μlpbs稀释至25μmol,储备待用。
测试方法:
1)配置母液,将化合物溶于dmso制成500μmol的母液,aβ1-42纤维母液制备方法同上;
2)以0.4mlpbs溶液为空白矫正仪器;
3)不同浓度的化合物1(终浓度0,5,10,20,30,40,60,80,100,200,500nm)溶于含有10%乙醇的pbs溶液,加入aβ1-42(终浓度1μmol)溶液至0.4ml,混合溶液在37℃条件下孵育30min,以589nm为激发光检测600–800nm的荧光发射光谱。取最大荧光发射波长641nm的数据用prism软件计算其kd值。
图2显示化合物1对aβ1-42纤维的kd值为19.1nm,表明化合物1对aβ蛋白具有高亲和力。
化合物2-30的亲和力测试方法同化合物1,测试结果如下表2。
表2化合物2-30与aβ1-42纤维亲和力测试
注:a表示kd值为1-10nm;b表示kd值为1-100nm;c表示kd值为100-500nm。
结果显示,化合物1-30对aβ1-42纤维均具有良好的亲和力,具有识别aβ的潜力。
测试例4:代表性化合物1的脑片荧光染色实验
实验方法:
12月龄app/ps1转基因鼠及其野生型小鼠麻醉,并用生理盐水灌注。取脑组织,在4%多聚甲醛中固定过夜,于20%、30%的蔗糖中梯度脱水过夜。包埋脑组织,在冰冻切片机中切片(20μm)。将脑切片与硫磺素s(ths)(10mg/ml)共孵育(每张脑片20μl),作为本实验aβ斑块染色的阳性对照。用50%乙醇洗涤3次后(每次洗涤1分钟),将该切片在室温下与化合物1(100μm)孵育20分钟。用无尘纸吸去残余液体后封片,在徕卡共聚焦显微镜下观察。
通过对12月龄app/ps1转基因小鼠的脑切片进行荧光染色,检测化合物1标识aβ斑块的能力。
共聚焦显微镜结果(图3)显示,大脑皮层和海马区均出现高对比度荧光斑点,通过与ths染色斑点比较,发现二者染色模式一致,证实这些斑点为aβ斑块。表明化合物1能够特异性识别脑切片上的aβ斑块。
测试例5:代表性化合物1的小动物活体成像实验
实验方法:采用ivis光谱成像系统(perkin/elmer)对化合物1的体内近红外成像能力进行评价。12月龄app/ps1转基因鼠及其野生型小鼠在成像前剃毛,异氟烷持续麻醉。拍摄零时间点照片后,转基因和野生型小鼠均注射2mg/kg新制备的化合物1(静脉注射,溶剂为5%dmso,5%cremophorel和90%生理盐水,0.4mg/ml)。连续记录脑内荧光信号,并利用活体成像软件对脑部区域荧光信号进行分析。
小动物活体荧光成像(图4)结果显示,小鼠尾静脉注射2.0mg/kg化合物1后,能快速有效区分转基因和野生型小鼠。表明化合物1具有活体识别阿尔茨海默病模型小鼠的能力。
测试例6:代表性化合物1的口服吸收实验
实验方法:
1)选择25%peg400,hs15,生理盐水作为口服给药溶剂,10mg/kg作为口服给药浓度,2mg/kg作为静脉给药浓度,对icr小鼠给药。选择15min、30min、1h、2h、4h、8h的时间点取血,3500rcf离心15min吸取上清得到血浆,生理盐水灌流取脑组织。
2)样品处理:在脑组织中加入pbs(4倍体积)匀浆,血浆稀释4倍,用有机溶剂(乙腈:水=1:1)萃取,离心吸取上清液检测。
3)lc-ms测试:以对照探针1为内标,建立化合物1的标准曲线(血浆线性范围50-2000ng/ml,脑组织线性范围50-2000ng/g),测定各时间点血浆和脑组织样品中化合物1的浓度。
如以下表3中所示,小鼠尾静脉给予化合物1(2mg/kg):探针快速清除,1h后血浆化合物1的浓度不足0.083h的1/10;0.083h脑组织中化合物1的浓度略低于血浆,为血浆的2/3。口服化合物1(10mg/kg)后,给药1h即可在组织上检测到高浓度的探针,显示化合物1口服后可快速吸收。以上结果表明化合物1口服可吸收,能够快速入脑,具有良好的生物相容性。
表3静脉和口服化合物1后的血浆和脑组织药物浓度
iv:静脉给药,po:口服给药,“\”:未进行该时间采血。
从以上测试例可以看出,本申请的化合物具有量子产率高、光学稳定性好、生物相容性优良的特点,并且其表现出与aβ蛋白高的亲和力,并且可以通过血脑屏障,因此可被用于阿尔茨海默病的体内诊断或监测。
- 还没有人留言评论。精彩留言会获得点赞!