一种烷基膦酸酯自催化水解制备烷基膦酸的方法与流程
1.本发明涉及一种烷基膦酸的制备方法,特别是涉及一种简单易行且能够准确判断反应终点、对反应装置无特殊要求、不使用强酸催化剂或脱酯化剂,以烷基膦酸酯为原料,经自催化水解反应制备烷基膦酸的方法。
背景技术:
2.有机膦酸及其衍生物是一类重要有机磷化工产品,其在矿物浮选、湿法冶金、阻燃剂、缓蚀剂、阻垢剂、纳米材料表面修饰以及药物合成等领域均具有重要作用。有机膦酸主要应用于溶剂萃取分离金属离子、缓蚀剂、阻垢剂以及自组装纳米材料的表面修饰。例如,1-辛基膦酸即为一种性能优异的铝镁缓蚀剂。二烷基膦酸则是一类性能优异的矿物浮选药剂,对稀土元素、锕系元素的金属离子具有良好的选择性。商品名为p507的(2-乙基己基)膦酸单(2-乙基己基)酯就是一种重要的湿法冶金萃取剂,广泛应用于铜-锌、镍-钴的分离,稀土元素的分组和单一稀土金属的分离提纯。此外,有机膦酸衍生物(金属盐类或胺盐)也是近年来开发的新一代磷系阻燃剂,可用于热塑性塑料(如pa、pbt)、纤维及纺织品的阻燃。
3.有机膦酸的分子结构如以下通式(i)所示,r为直链或支链c1~c20的烷基,或者为直链或支链c7~c20的芳烷基。
[0004][0005]
有机膦酸的制备方法较多。比较常见的是先以arbuzov反应制备相应的有机膦酸酯,然后在强酸或强碱存在条件下,经加热水解而得到有机膦酸。如专利cn 105503944采用浓硫酸和浓盐酸对烷基膦酸酯进行水解;专利cn103384675使用硫酸或杂多酸等不挥发性酸为催化剂水解烷基膦酸酯;专利cn105153224则以氢溴酸为催化剂水解烷基膦酸酯。但是,这些方法由于使用了强酸,须使用耐强酸腐蚀的特殊反应设备,而且后处理还须脱除强酸催化剂,存在“三废”排放多的缺点。
[0006]
claudia ehrhardt等人则以三甲基溴硅烷为烷基膦酸酯的脱酯化剂制备烷基膦酸,但这种方法的缺点是价格较高,后处理工艺复杂,而且收率偏低。
[0007]
近年来,科莱恩公司专利(cn 104093724)公开了一种以烯烃为原料,与次磷酸加成生成烷基次膦酸,然后经强氧化剂氧化、盐酸酸化直接制得烷基膦酸的方法。其反应式如下:
[0008][0009]
这种方法虽然反应原料来源广泛,但钯系贵金属催化剂pd2dba3[三(二亚苄基丙
酮)二钯(0)]和偶联剂xantphos[4,5-双(二苯基膦)-9,9-二甲基氧杂蒽]均非常昂贵,而且该路线易产生烯烃二聚后与次磷酸加成的副产物。此外,该反应工艺看似简单,实则流程复杂,须除去催化剂、无机盐,并反复重结晶才能得到目标产物,工业化生产成本高昂。
[0010]
在欧洲专利ep0529640a中公开了一种烷基膦酸二烷基酯的自催化水解方法。该方法于常压下在反应器中加入一定量的烷基膦酸二烷基酯再加入占该烷基膦酸二烷基酯5~20wt%的烷基膦酸作为催化剂;将上述反应体系加热至160~210℃后,以控制反应混合物温度不低于160℃的速率滴加不超过化学计量的水直至反应结束,作为反应物的水以少量不连续或连续的方式加入;反应器上方设置的冷凝器以温度高于待除去的醇(即,水解释放的醇)沸点5℃的冷却介质进行冷凝,从而使醇能够从冷凝器中逸出进而在冷凝器末端被冷凝收集,而水无法逸出,进而回流至反应体系继续参与水解反应。当醇的馏出量达到理论量时,反应完成,将反应混合物冷却至110~120℃,减压除去残留的水和醇,从而得到糖浆状的烷基膦酸。在这种方法中,不可避免地会存在以下几个问题:(1)该烷基膦酸二烷基酯的自催化水解方法是以定量水
--“
不超过化学计量的水”进行水解的方法。所谓“化学计量的水”即“在投料量的烷基膦酸二烷基酯水解所需的理论水量基础上略微过量”得来(ep0529640a第3栏第13行至19行;实施例1、2和3)。理论上,虽然可以通过在冷凝器中使用高温冷却介质(高于水解产生的低级醇沸点5℃)实现水能够冷凝回流继续参与反应、低级醇能逸出反应体系的目的,但实际上,由于反应体系的温度高于160℃,汽化水有强烈逸出反应体系的趋势,更重要的是水与低级醇形成共沸物后,汽化水不可避免会与低级醇一并逸出反应体系,必然造成参与水解反应的水量低于化学计量的水,由此必然造成水解反应不彻底的现象。(2)为确保烷基膦酸二烷基酯有较高的水解程度,必须尽量使得加入的水几乎全部参与烷基膦酸二烷基酯的水解,因此该方法对冷凝系统和冷却介质有特殊要求,才能使得汽化的水回流到体系继续参与水解反应,同时水解产生的副产物低级醇又能不被冷凝回流而从反应体系逸出,从而促使水解反应正向移动。但这在实际实施时存在较大困难,如果采用实际工业装置中通常使用的循环水冷却塔作为冷却系统,其温度无法调控,也无法升至高于70℃的水平;当需要冷却共沸点接近100℃的水与低级醇形成的共沸物时,必须使用非水的冷却介质且控制非水冷却介质的温度高于醇的沸点5℃,但由于共沸现象的存在,仍无法保证醇能彻底逸出,水能不逸出而全部返回反应体系。(3)这种方法对反应终点的判断是以副产物低级醇的馏出量达到理论量为标准的,但如前所述,由于低级醇与水形成共沸物,冷却系统很难百分百地确保达到“汽化水回流系统,而低级醇彻底逸出”的理想状态,因此以“当化学计量的水被滴加至反应体系,副产物低级醇气流基本停止”(第四栏第8行至第10行、第四栏第34行至第35行、第四栏第56行至第57行)为反应终点进行判断时,并不能准确判断水解反应的程度和反应终点。
技术实现要素:
[0011]
本发明的目的在于克服现有技术的不足之处,提供一种简单易行且能够准确判断反应终点、对反应装置无特殊要求、不使用强酸催化剂或脱酯化剂,以烷基膦酸酯为原料,经自催化水解反应制备烷基膦酸的方法。
[0012]
本发明所要解决的技术问题通过以下技术方案予以实现:
[0013]
本发明提供一种烷基膦酸酯自催化水解制备烷基膦酸的方法,所述方法包括:
[0014]
将由以下通式(i)所示的烷基膦酸作为催化剂加入作为原料的由以下通式(ii)所示的烷基膦酸酯中,所述催化剂的加入量为所述原料的重量的1wt%~15wt%,将得到的反应混合物加热至150℃~200℃;
[0015]
在150℃~200℃,缓慢且持续地向所述反应混合物中加入蒸馏水,进行自催化水解反应;所述水解反应产生的副产物低级醇和被汽化的水经由冷凝器冷却后,在分水接收器收集;
[0016]
以核磁磷谱或氢谱实时监测作为原料的所述烷基膦酸酯的特征峰,来判断所述水解反应进行的程度,即,待所述烷基膦酸酯的特征核磁磷谱峰或氢谱峰消失,则达到烷基膦酸酯水解完全的反应终点,停止向所述反应混合物中加入蒸馏水;
[0017]
真空脱除所述反应混合物中残留的水分和副产物低级醇,得到由以下通式(i)所示的烷基膦酸;
[0018][0019]
其中,在通式(i)和通式(ii)中,r为直链或支链的c1~c20的烷基,或者为直链或支链的c7~c20的芳烷基,r1和r2为相同或不同的c1~c8的烷基。
[0020]
在本发明的实施方式中,所述的催化剂为如通式(i)所示的目标烷基膦酸自身,无需其它类型催化剂。所述催化剂的加入量为所述原料的重量的1wt%~15wt%,优选5wt%~10wt%。
[0021]
在本发明的实施方式中,所述蒸馏水是在保持反应体系温度始终在150℃~200℃的条件下,持续缓慢加入反应体系,直至达到所述烷基膦酸酯的水解完全的反应终点,才停止加入蒸馏水。例如,蒸馏水可通过计量泵或蠕动泵缓慢引入反应体系。
[0022]
在本发明的实施方式中,副产物低级醇及汽化的水经由循环水冷凝器冷却后,在分水接收器收集,不会回流进入反应体系。
[0023]
在本发明的实施方式中,所述水解反应的进程是以核磁磷谱或氢谱实时监测原料烷基膦酸酯的特征核磁磷谱峰或氢谱峰,待该特征峰消失,则判定反应终点。
[0024]
在本发明的实施方式中,在自催化水解反应结束后,真空脱除残留的水分和副产物低级醇,即得如通式(i)所示的烷基膦酸产物(纯度高于98%)。进一步重结晶后,可使烷基膦酸的产物纯度高于99%。
[0025]
与现有技术相比,本发明具有如下有益效果:
[0026]
(1)本发明的制备方法不使用传统的浓硫酸、浓盐酸、氢溴酸等强酸催化剂或三甲基溴硅烷等脱酯化剂,仅使用目标烷基膦酸为催化剂,通过自催化水解相应烷基膦酸酯得到烷基膦酸产物。该制备方法不仅对设备要求较低,无需耐强酸腐蚀的特殊材质反应器或复杂设备,简单的反应釜即可满足要求,而且后处理工艺简单,无需脱除催化剂,也不产生酸性催化剂废弃物,具有显著的环保特征。
[0027]
(2)本发明的制备方法工艺简单,反应条件温和,水解反应彻底,所得烷基膦酸产物纯度高(纯度高于98%),无需复杂后处理纯化工艺,适宜工业化生产。
[0028]
(3)与现有的烷基膦酸二烷基酯的自催化水解方法(例如ep0529640a)相比,本发明的方法是以实时取样监测反应体系的原料烷基膦酸酯的核磁磷谱或氢谱,待其特征核磁磷谱峰或氢谱峰消失为反应终点,由此可以准确判断出烷基膦酸酯水解反应的终点;而且,本发明的方法中,水的用量以最终烷基膦酸二烷基酯达到完全水解的程度而定,即检测到烷基膦酸二烷基酯完全水解后才停止加入水,所以在水解反应进行期间是持续向反应体系缓慢加入水(只需控制蒸馏水的进料速度使得反应体系温度不低于150℃即可),汽化的水无需返回反应体系继续参加反应,因此水被汽化逸出并不影响水解反应的彻底性;也正因此,本发明对于冷却系统及冷却介质温度的要求并不苛刻,相较于ep0529640a的方法,本发明的方法中冷凝器只需常规冷却水起循环冷却作用即可,无需温度可调节或可控制的特殊冷却系统和特殊冷却介质。
附图说明
[0029]
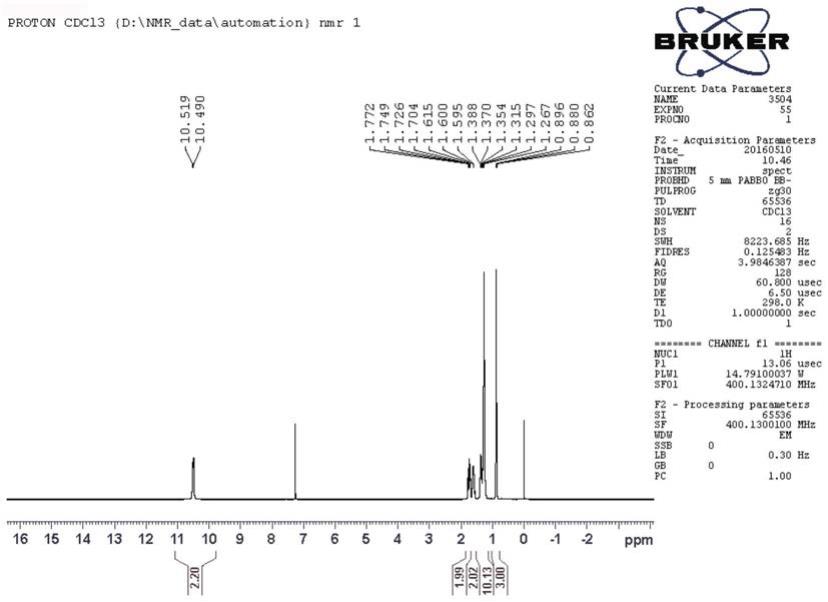
图1是实施例1制备的1-辛基膦酸产品的核磁氢谱图;
[0030]
图2是实施例1制备的1-辛基膦酸产品的核磁磷谱图;
[0031]
图3是实施例2制备的1-十二烷基磷酸的核磁氢谱图;
[0032]
图4是实施例2制备的1-十二烷基磷酸的核磁磷谱图;
[0033]
图5是实施例3制备的1-十六烷基磷酸的核磁氢谱图;
[0034]
图6是实施例3制备的1-十六烷基磷酸的核磁磷谱图。
具体实施方式
[0035]
实施例1
[0036]
将200g 1-辛基膦酸二乙酯和15g 1-辛基膦酸的混合物在装配有机械搅拌器、温度计、蠕动泵进料器和分水蒸馏接收管的四口圆底烧瓶中搅拌加热至190℃。到达该温度后,开启蠕动泵,缓慢向上述反应体系加入蒸馏水。蒸馏水进样速度必须控制在能保证反应体系温度不低于180℃为宜。同时在分水蒸馏接收管收集副产物乙醇。反应进程以实时取样进行核磁磷谱分析监测,反应终点以原料1-辛基膦酸二乙酯的特征核磁磷谱峰
31
p nmr(162mhz,cdcl3,δ32.43)消失,全部转化为目标产物1-辛基膦酸的特征核磁磷谱峰
31
p nmr(162mhz,cdcl3,δ37.63)进行判定。自催化水解反应结束后真空脱除残留水分和少量乙醇,即得目标产物1-辛基膦酸(纯度99.4%,熔点99.2~103.3℃)。
[0037]
实施例2
[0038]
将200g 1-十二烷基膦酸二乙酯和10g 1-十二烷基膦酸的混合物在装配有机械搅拌器、温度计、蠕动泵进料器和分水蒸馏接收管的四口圆底烧瓶中搅拌加热至190℃。到达
该温度后,开启蠕动泵,缓慢向上述反应体系加入蒸馏水。蒸馏水进料速度必须控制在能保证反应体系温度不低于180℃为宜。同时在分水蒸馏接收管收集副产物乙醇。反应进程以实时取样进行核磁磷谱分析监测,反应终点以原料1-十二烷基膦酸二乙酯的特征核磁磷谱峰
31
p nmr(162mhz,cdcl3,δ32.34)消失,全部转化为目标产物1-十二烷基膦酸的特征核磁磷谱峰
31
p nmr(162mhz,cdcl3,δ38.31)进行判定。自催化水解反应结束后真空脱除残留水分和少量乙醇,即得目标产物1-十二烷基膦酸(纯度为99.2%,熔点100.9℃~103.3℃)。
[0039]
实施例3
[0040]
将200g 1-十六烷基膦酸二丁酯和20g 1-十六烷基膦酸的混合物在装配有机械搅拌器、温度计、蠕动泵进料器和分水蒸馏接收管的四口圆底烧瓶中搅拌加热至190℃。到达该温度后,开启蠕动泵,缓慢向上述反应体系加入蒸馏水。蒸馏水进料速度必须控制在能保证反应体系温度不低于180℃为宜。同时在分水蒸馏接收管收集副产物正丁醇。反应进程以实时取样进行核磁磷谱分析监测,反应终点以原料1-十六烷基膦酸二丁酯的特征核磁磷谱峰
31
p nmr(162mhz,thf-d8,δ32.26)消失,全部转化为目标产物1-十六烷基膦酸的特征核磁磷谱峰
31
p nmr(162mhz,thf-d8,δ30.74)进行判定。自催化水解反应结束后真空脱除残留水分和少量丁醇,即得目标产物1-十六烷基膦酸(纯度为99.3%,熔点96.8~98.5℃)。
[0041]
本发明的制备方法仅以少量目标烷基膦酸为催化剂,制备工艺简单,无需复杂后处理工艺,对设备要求低,不产生酸性废弃物,具有显著的环保特征,且水解反应彻底,所得烷基膦酸产物纯度高,适宜工业化生产。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1