本发明涉及分子生物学技术领域,具体涉及一种基于转录组序列开发多态性ssr标记的方法。
背景技术:
ssr标记,又称为微卫星dna,是一种基于特异性引物pcr扩增的分子标记,由2到6个核苷酸序列重复基序组成。其标记两端的序列较为保守,根据这一保守区域设计引物,通过pcr技术实现ssr位点扩增,可直接反映不同样本或物种间该dna片段的遗传多态性。鉴于ssr标记多态性丰富且随机分布于基因组中,目前已被广泛应用于基因的快速定位、指纹图谱构建以及分子标记辅助选择育种等研究。
按照其来源,ssr标记分为基因组(genome)ssr标记和转录组ssr标记两种类型。转录组ssr标记相对于基因组ssr标记具有以下优点:(1)对于基因组序列未知的物种而言,开发成本低廉,高效;(2)由于转录组序列的种间保守性强,转录组ssr标记可在相近的种属中使用,为生物进化和系统发育学的研究提供工具;(3)可与特定功能的基因紧密连锁,更易与表型性状相联系。随着高通量测序(ngs)技术的发展,低廉的价格即可获得大量转录组数据,而从其中的表达序列开发出来的ssr位点也得到了广泛的应用,目前应用于遗传图谱的构建、关联分析、遗传多样性的评价、种质资源的鉴定和系统进化学等研究。
可见,筛选并利用转录组ssr在分子生物学研究中具有重要地位。现有基于转录组序列开发ssr的方法,效率较低,需要先通过分子生物学实验验证其可扩增性,进一步筛选多态性标记以供使用;同时,现有多态性ssr标记开发方法,多基于测序原始数据进行筛选,计算量偏大,对电脑要求较高。针对以上不足,本方法以转录组序列为对象,普通电脑即可实现样本间共有ssr标记鉴定与多态性标记筛选,同时,增加了获得ssr标记的真实性与多态性效率。目前,还未见有关此方法的相关报道。
技术实现要素:
本发明提供一种基于转录组序列开发多态性ssr标记的方法,有效解决多态性ssr标记的常规筛选方法效率低,计算量大,可操作性差,对计算机配置依赖程度高的问题。
为实现上述目的,本发明采用以下技术方案实现:
一种基于转录组序列开发多态性ssr标记的方法,包括以下步骤:
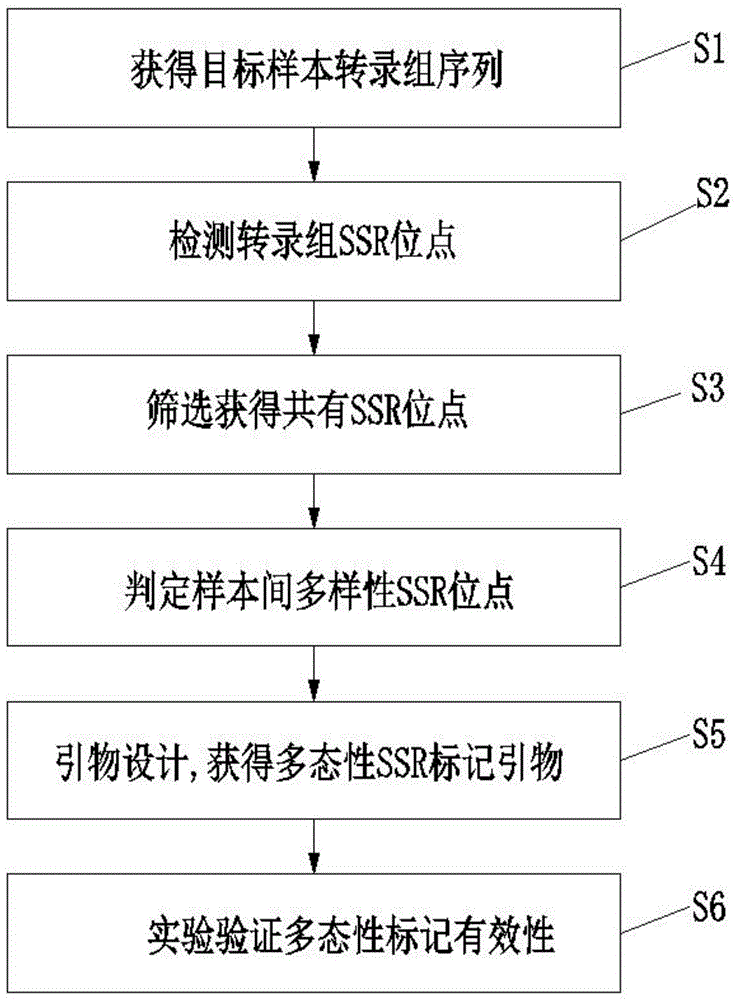
s1、获得目标物种多个样本的转录组序列;
s2、对每个样本转录组序列,分别检测潜在的ssr位点信息;
s3、对步骤s2中检出的ssr位点信息,利用序列筛选程序进行筛选,获得ssr位点重复基序类型与其相邻核苷酸序列信息;
s4、对步骤s3中检出的ssr位点重复基序与其核苷酸序列信息,采用perl语言编写程序,从中筛选样本间多态性ssr候选位点;
s5、对步骤s4中携带多态性ssr位点的转录组序列集,拼接为无重复的叠连群uni-contig,依据其侧翼保守序列设计引物,进行标记多态性检测。
作为上述方案的优选,还包括以下步骤s6:利用获得的ssr标记引物对,以目标物种的基因组dna为模板,分别执行pcr扩增,根据扩增结果验证引物的有效性。
作为上述方案的优选,还包括以下步骤s7:对uni-contig序列,搜索ncbi蛋白质数据库,依据e-value最小原则判定与ssr位点连锁的基因功能。
作为上述方案的优选,在步骤s1中,首先取目标物种的若干样本,分别对其转录组进行高通量测序获得reads,再通过trinity软件将测序得到的所有reads进行拼接组装,最终获得转录组序列。
作为上述方案的优选,在步骤s2中,转录组序列进行ssr位点搜索,其中,二碱基、三碱基、四碱基、五碱基与六碱基基序重复次数分别至少为6、5、4、4、4次。
作为上述方案的优选,在步骤s3中,与ssr位点重复基序相邻的上、下游核苷酸序列长度设为10bp。
作为上述方案的优选,在步骤s4中,依据ssr重复基序与相邻10bp核苷酸序列一致,判定为相同ssr位点;以重复基序存在变异,判定为多态性ssr候选标记。
作为上述方案的优选,在步骤s5中,根据引物在uni-contig中的跨度区间,准确计算该ssr标记pcr扩增产物长度。
由于具有上述结构,本发明的有益效果在于:
本申请提供了一种基于转录组序列开发多态性ssr标记的方法,该方法首先获取目标物种多个样本转录组数据;鉴定每个样本ssr位点信息;依据ssr重复基序及其相邻核苷酸序列,判定样本间共有ssr位点;若样本间ssr位点重复基序存在变异则视为多态性ssr位点;进一步根据非冗余uni-contig序列设计引物,预测pcr扩增片段长度及与其连锁基因的功能;上述筛选结果利用pcr扩增验证其有效性;
本申请形成了一套严格有效的ssr标记开发方法,能够利用转录组序列有效筛选多态性的ssr标记,降低对计算机的配置要求,普通电脑即可完成,有效提高分子标记引物的有效性,缩短开发时间与成本,同时还可预测多态性ssr标记的pcr扩增片段长度及其连锁基因功能信息,显著提高转录组ssr标记的开发效率,促进分子辅助育种与功能基因研究。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍。
图1为本发明的方法流程框图;
图2为本发明多态性ssr标记引物扩增产物的聚丙烯酰胺凝胶电泳检测图。
具体实施方式
下面将结合本发明的附图,对本发明的技术方案进行清楚、完整地描述。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
如图1、图2,本申请提供一种基于转录组序列开发多态性ssr标记的方法,包括以下步骤:
s1、首先取目标物种的若干样本,分别对其转录组进行高通量测序获得reads,去除低质量的测序read(质量值q≤5的碱基数占整个read的50%以上),以确保获得高质量转录组序列,再通过trinity软件将测序得到的所有reads进行拼接组装,最终获得目标物种多个样本的转录组序列;
s2、对步骤s1获得的每个样本转录组序列,分别利用misa软件检测出潜在的ssr位点信息,利用misa软件鉴定ssr位点,应满足以下条件:对于二核苷酸重复基序,重复次数设为≥6;对于三核苷酸重复基序,重复次数设为≥5;对于四、五、六核苷酸重复基序,重复次数设为≥4;
s3、对步骤s2中检出的ssr位点信息,利用seq_motif.pl脚本,获得ssr位点重复基序类型与其相邻核苷酸序列信息,筛选出在两个样本中均存在(共有)的ssr位点,其中,与ssr位点重复基序相邻的上、下游核苷酸序列长度设为10bp;
s4、对步骤s3中检出的共有ssr位点,进一步执行poly_motif.pl程序,获得多态性ssr位点,具体的,依据ssr重复基序与相邻10bp核苷酸序列一致,判定为相同ssr位点;以重复基序存在变异,判定为多态性ssr候选标记;
s5、对步骤s4中携带多态性ssr位点的转录组序列集,利用clcgenomicsworkbench软件拼接为非冗余叠连群uni-contig(拼接软件可以但不限于clcgenomicsworkbench,其他同类拼接软件也可以),依据其侧翼保守序列设计引物(可以使用primer3.0_core.exe软件,进行引物批量设计),进行标记多态性检测,得到多态性引物,而且,根据引物在uni-contig中的跨度区间,能够准确计算该ssr标记pcr扩增产物长度。
还包括以下步骤s6:利用获得的ssr标记引物对,以目标物种的基因组dna为模板,分别执行pcr扩增,根据扩增结果验证引物的有效性。
还包括以下步骤s7:对uni-contig序列,搜索ncbi蛋白质数据库,依据e-value最小原则判定与ssr位点连锁的基因功能。
现以魔芋为例,对转录组多态性ssr标记的开发与验证过程进行举例说明。
步骤一、对魔芋进行转录组测序:对珠芽黄魔芋与白魔芋,利用植物总rna提取试剂盒(天根生化科技有限公司)分别进行rna提取,用带有oligo(dt)的磁珠富集真核生物,加入fragmentationbuffer,将mrna打断成短片段,以mrna为模板,用六碱基随机引物合成第一条cdna链,然后加入缓冲液、dntps、rnaseh和dnapolymerasei合成第二条链,在经过qiaquickpcr试剂盒纯化并加eb缓冲液洗脱之后做末端修复、加poly(a)并连接测序头,然后用琼脂糖凝胶电泳进行片段大小选择最并连接测序头,建好的测序文库用扩增,建好的测序文库用illuminahiseqtm2000进行测序。最终从珠芽黄魔芋中获得13599908条测序rawreads,从花魔芋中获得15867314条测序rawreads。
步骤二、拼接获得转录组序列:将测序获得的原始转录组数据(rawreads),去除接头序列及低质量reads,利用trinity软件进行无参拼接组装,白魔芋和珠芽黄魔芋分别组装得到54596条、58858条contigs,平均长度为697.5bp、618.3bp,n50分别是1239bp和1127bp。白魔芋最大长度12002bp,最小长度201bp;珠芽黄魔芋最长8330bp,最短201bp。
步骤三、样本ssr位点信息获取:对每个样本转录组contigs序列,利用misa软件识别出ssr位点。搜索参数:2-6个核苷酸重复基序,最小重复次数分别为6,5,4,4和4。在珠芽黄魔芋中,鉴定到7809条序列,共包含8083个ssr位点;其中1169条contigs携带≥2个微卫星位点。在白魔芋中,鉴定了8757条序列,共包含9074个微卫星位点;其中1420条contigs携带≥2个微卫星位点。
步骤四、样本间相同(共有)ssr位点鉴定:对步骤二中每个样本转录组序列与步骤三)中ssr位点信息,执行seq_motif.pl程序。在两个样本间,共检测到298个共有微卫星位点。在珠芽黄魔芋与白魔芋中,共有标记的百分比分别是3.83%和3.41%,共有标记比例较低,这与本实例选用的是亲缘关系较远的两个物种一致。
步骤五、多态性ssr位点计算:执行poly_motif.pl脚本,判定ssr基序变异信息,获得样本间重复基序存在变异的ssr位点。从298个共有微卫星位点中,筛选到141个多态性标记,多态性比例达47.3%。
步骤六、多态性引物设计:对携带多态性ssr位点contig序列,利用clcgenomicsworkbench的uni-contig序列,利用primer3.0_core.exe进行批量引物设计,最终成功获得112对电子多态性引物,如表1所示,成功率达79.43%。
表1珠芽黄魔芋与白魔芋间电子多态性引物列表
步骤七、多态性引物验证:取珠芽黄魔芋与白魔芋嫩叶,利用ctab法提取其基因组dna,0.8%琼脂糖凝胶电泳检测其完整性;本申请随机对其中的40对引物进行pcr扩增验证,并进一步利用6%丙烯酰胺凝胶电泳检测其pcr产物多态性。其中21对引物可以在珠芽黄魔芋、白魔芋中均获得扩增条带,其中多态性引物数为16,多态性比例为76.2%。
本实施例中,目标样本珠芽黄魔芋与白魔芋,属于两个物种,ssr标记可转移率较低,若利用常规方法进行多态性ssr筛选,检测效率更低。通过本实施例,从转录组数据中获得多态性ssr标记,然后利用pcr扩增进行验证,有效提高了多态性ssr筛选的效率。
其中,软件misa、primer3.0_core.exe均为分子生物学领域常规软件,clcgenomicsworkbench由qiagen公司出品的测序数据分析工具,其使用方法可依据本领域的一般技术常识实施。所述的perl脚本seq_motif.pl和poly_motif.pl,由本申请自主编写,其具体代码见如下。
(1)perl脚本seq_motif.pl,其代码如下:
(2)perl脚本poly_motif.pl,其代码如下:
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。