一种铂配合物及其应用、包含其的有机光电装置的制作方法
本发明属于光电领域,涉及一种铂配合物及其应用、包含其的有机光电装置。
背景技术:
能够吸收和/或发光的化合物可适用于各种光学和光电器件,包括但不限于诸如太阳能、光敏这样的光吸收器件、有机发光二极管(oled)、光发射器件或既有光吸收又有光发射能力的器件以及用于生物标记的相关应用。为了发现用于光学和电致发光器件的有机和有机金属材料,本领域内已专门作过很多研究。可应用于发光和照明器件的光电材料(红色和绿色有机金属材料用作磷光材料,蓝色有机金属材料用作荧光材料)的研究取得了显著的进步,并且已在有机发光二极管(oled)照明和先进显示器的应用中取得成功。不过,目前在大尺寸显示设备的应用上还存在发光寿命短、发热量大和实际效率不高等缺点。
一般认为,400~450nm的短波长蓝光(高能量蓝光)为对于眼睛伤害最大,可以产生数码视觉疲劳和影响睡眠,最终导致近视、白内障以及黄斑病变的眼睛病理危害和人体节律危害。设计发光区间在450-500nm之间的蓝光光源并应用到相关的电子产品,可以从根源上解决如今电子设备中的高能量蓝光对人体伤害问题。
然而,相比于红色和绿色发光材料而言,优良的蓝光发光材料更为稀少,尤其是同时具有稳定结构和合适的发光光谱的高效率磷光蓝光材料分子,具有很大的需求。与红色及绿色磷光体的最低三重激发态能量相比,蓝色磷光体的最低三重激发态能量很高,这意味着蓝色器件的主体材料的最低三重激发态能量必须更高。因此,能达到蓝光发光区间的有机光电装置类型较为有限,相应地,要调控合适的蓝光光谱、使得蓝色发光器件在发光过程中表现出优秀的性能则更为困难。
技术实现要素:
针对现有技术的不足,本发明的目的之一在于提供一种铂配合物,所述铂配合物包括如式a所示的结构片段:
其中,re选自取代或未取代的c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷基、取代或未取代的c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)芳基中的任意一种;
rd和rf独立地表示单取代基到最大允许取代基,并且各自独立地选自氢原子、氕原子、氘原子、卤素、取代或未取代的c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷基、取代或未取代的c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)烯基、取代或未取代的c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)炔基、取代或未取代的c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷氧基、氨基、羟基、巯基、硝基、氰基、异氰基、亚硫酰基、磺酰基、羧基、肼基、芳氧基、酯基、烷氧基羰基、酰胺基、烷氧基羰基氨基、芳氧基羰基氨基、氨磺酰基、氨基甲酰基、烷硫基、脲基、磷酰胺基、甲硅烷基、取代或未取代的c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)芳基、取代或未取代的c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)芳基氨基、取代或未取代的c3-c30(例如c4、c6、c8、c12、c15、c18、c20、c23、c25、c28等)杂芳基、聚合物基团中的任意一种;其中聚合物基团包括聚烯烃基、聚醚、聚酯和含有重复单元的其它组,示例性地包括但不限于,-(ch2o)n-ch3、-(ch2ch2o)n-ch3、-[ch2ch(ch3)]n-ch3、-[ch2ch(cooch3)]n-ch3、-[ch2ch(cooch2ch3)]n-ch3和-[ch2ch(cootbu)]n-ch3,其中n为大于等于1的整数。
式a中,
式a中,
当上述基团存在取代基时,所述取代基各自独立地选自氘、氚、氰基、卤素、c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)的烷基、c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)烯基、c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)炔基、c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷氧基、氨基、芳氧基、酯基、烷氧基羰基、酰胺基、烷氧基羰基氨基、芳氧基羰基氨基、氨磺酰基、氨基甲酰基、烷硫基、脲基、磷酰胺基、甲硅烷基、c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)的芳基、c3-c30(例如c4、c6、c8、c12、c15、c18、c20、c23、c25、c28等)的杂芳基中的任意一种。
本发明中,杂芳基中的杂原子通常是指n、o或s;
本发明提供了一种新型的配合物,包括式a片段,即将甲基吡啶并咪唑型卡宾结构螯合配位,使得到的配合物具有较高的稳定性和发光效率,其中偶然发现吡啶咪唑片段上吡啶n对位的甲基有明显提高发光效率的作用;相关理论原因之一可能在于卡宾结构具有合适的三重态能量并且碳铂配位键相较于氮铂配位键更稳定,因此该配合物具有较好的稳定性;相关理论原因之二可能在于甲基吡啶并咪唑结构能够增加配体中激发态部分的π体系,致使整个光谱可以变窄,这将能够促进发射光色并改善器件的性能;相关理论原因之三可能在于,能够影响lumo轨道的能级,从而影响三线态的激态分布,从而极大的提高了铂配合物的发光效率。
优选地,所述铂配合物具有式i所示的结构:
式i中,ra、rb、rc、rd和rf独立地表示单取代基到最大允许取代基,并且各自独立地选自氢原子、氕原子、氘原子、卤素、取代或未取代的c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷基、取代或未取代的c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)烯基、取代或未取代的c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)炔基、取代或未取代的c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷氧基、氨基、羟基、巯基、硝基、氰基、异氰基、亚硫酰基、磺酰基、羧基、肼基、芳氧基、酯基、烷氧基羰基、酰胺基、烷氧基羰基氨基、芳氧基羰基氨基、氨磺酰基、氨基甲酰基、烷硫基、脲基、磷酰胺基、甲硅烷基、取代或未取代的c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)芳基、取代或未取代的c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)芳基氨基、取代或未取代的c3-c30(例如c4、c6、c8、c12、c15、c18、c20、c23、c25、c28等)杂芳基、聚合物基团中的任意一种;
式i中,re选自取代或未取代的c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷基、取代或未取代的c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)芳基中的任意一种;
当上述基团存在取代基时,所述取代基各自独立地选自氘、氚、氰基、卤素、c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)的烷基、c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)烯基、c2-c10(例如c3、c4、c5、c6、c7、c8、c9等)炔基、c1-c10(例如c2、c3、c4、c5、c6、c7、c8、c9等)烷氧基、氨基、芳氧基、酯基、烷氧基羰基、酰胺基、烷氧基羰基氨基、芳氧基羰基氨基、氨磺酰基、氨基甲酰基、烷硫基、脲基、磷酰胺基、甲硅烷基、c6-c30(例如c10、c12、c14、c16、c18、c20、c26、c28等)的芳基、c3-c30(例如c4、c6、c8、c12、c15、c18、c20、c23、c25、c28等)的杂芳基中的任意一种。
本发明提供了一种新型的如式i所示的配合物,通过在甲基吡啶并咪唑型卡宾结构中引入二价铂配合物配体,该配合物具有较高的稳定性和发光效率。
本发明中该配合物易于制备和升华提纯,溶于一般有机溶剂,能同时适合蒸镀法和溶液法加工的器件制程。
优选地,所述ra、rb、rc、rd和rf各自独立地选自氢原子、氕原子、氘原子、卤素、取代或未取代的c1-c10烷基、取代或未取代的c6-c30芳基中的任意一种;
优选地,所述卤素包括氟原子、氯原子、溴原子或碘原子中的任意一种;
优选地,所述取代或未取代的c1-c10烷基包括甲基、苯甲基、二苯基甲基、三苯基甲基、乙基、2-苯基乙基、2,2-苯基乙基、2,2,2-三氟乙基、丙基、异丙基、3,3,3-三氟丙基、1,1,1,3,3,3-六氟-2-丙基、丁基、异丁基、六氟异丁基、叔丁基;环丙基、环丁基、环戊基、环己基或环庚基中的任意一种;
优选地,所述取代或未取代的c6-c30芳基包括苯基、2-甲基苯基、2-异丙基苯基、2-乙基苯基、4-甲基苯基、4-异丙基苯基、4-乙基苯基、4-叔丁基苯基、2,3-二甲基苯基、2,3-二乙基苯基、2,3-二异丙基苯基、2,3-二异丁基苯基、2,3-二环己基苯基、2,3-二环丙基苯基、2,3-二环丁基苯基、2,3-二环戊基苯基、2,4-二甲基苯基、2,4-二乙基苯基、2,4-二异丙基苯基、2,4-二异丁基苯基、2,4-二环己基苯基、2,4-二环丙基苯基、2,4-二环丁基苯基、2,4-二环戊基苯基、2,6-二甲基苯基、2,6-二乙基苯基、2,6-二异丁基苯基、2,6-二环己基苯基、2,6-二环丙基苯基、2,6-二环丁基苯基、2,6-二环戊基苯基、3,5-二甲基苯基、3,5-二乙基苯基、3,5-二异丙基苯基、3,5-二异丁基苯基、3,5-二环己基苯基、3,5-二环丙基苯基、3,5-二环丁基苯基、3,5-二环戊基苯基、2,3,5,6-四甲基苯基、2,4,6-三甲基苯基、2,4,6-三乙基苯基、2,4,6-三异丙基苯基、2,4,6-三异丁基苯基、2,4,6-三环己基苯基、2,4,6-三环丙基苯基、2,4,6-三环丁基苯基或2,4,6-三环戊基苯基中的任意一种。
优选地,所述ra、rb、rc、rd和rf各自独立地选自氢、甲基、异丙基、2,6-二甲基苯基、2,4,6-三甲基苯基或2,6-二异丙基苯基中的任意一种。
优选地,所述re选自甲基、苯甲基、二苯基甲基、三苯基甲基、乙基、2-苯基乙基、2,2-苯基乙基、2,2,2-三氟乙基、丙基、异丙基、3,3,3-三氟丙基、1,1,1,3,3,3-六氟-2-丙基、丁基、异丁基、六氟异丁基、叔丁基、环丙基、环丁基、环戊基、环己基、环庚基、苯基、2-甲基苯基、2-异丙基苯基、2-乙基苯基、4-甲基苯基、4-异丙基苯基、4-乙基苯基、4-叔丁基苯基、2,3-二甲基苯基、2,3-二乙基苯基、2,3-二异丙基苯基、2,3-二异丁基苯基、2,3-二环己基苯基、2,3-二环丙基苯基、2,3-二环丁基苯基、2,3-二环戊基苯基、2,4-二甲基苯基、2,4-二乙基苯基、2,4-二异丙基苯基、2,4-二异丁基苯基、2,4-二环己基苯基、2,4-二环丙基苯基、2,4-二环丁基苯基、2,4-二环戊基苯基、2,6-二甲基苯基、2,6-二乙基苯基、2,6-二异丁基苯基、2,6-二环己基苯基、2,6-二环丙基苯基、2,6-二环丁基苯基、2,6-二环戊基苯基、3,5-二甲基苯基、3,5-二乙基苯基、3,5-二异丙基苯基、3,5-二异丁基苯基、3,5-二环己基苯基、3,5-二环丙基苯基、3,5-二环丁基苯基、3,5-二环戊基苯基、2,3,5,6-四甲基苯基、2,4,6-三甲基苯基、2,4,6-三乙基苯基、2,4,6-三异丙基苯基、2,4,6-三异丁基苯基、2,4,6-三环己基苯基、2,4,6-三环丙基苯基、2,4,6-三环丁基苯基或2,4,6-三环戊基苯基中的任意一种。
优选地,所述re选自甲基、异丙基、2,6-二甲基苯基或2,4,6-三甲基苯基中的任意一种。
优选地,所述铂配合物具有如下(iii-1)-(iii-8)所述的结构中的任意一种:
其中,ra、rb、rc具有与前文相同的限定范围。
优选地,所述ra、re和rf各自独立地选自甲基、乙基、异丁基、叔丁基、异丁基、2,6-二甲基苯基、2,6-二乙基苯基、2,6-二异丙基苯基、2,6-二异丁基苯基、2,4,6-二甲基苯基、2,4,6-二乙基苯基、2,4,6-二异丙基苯基、2,4,6-二异丙基苯基或2,4,6-二异丁基苯基;
优选地,所述rb、rc和rd各自独立地选自甲基。
优选地,所述铂配合物具有如下配合物1-配合物228所示结构中的任意一种:
本发明的目的之二在于提供一种如目的之一所述的铂配合物的应用,所述铂配合物应用于有机光电装置。
优选地,所述铂配合物用作有机光电装置的磷光发光材料;
优选地,所述铂配合物用作有机光电装置的蓝磷光发光材料;
优选地,所述铂配合物用作有机光电装置的窄光谱发光材料。
在本发明中,铂配合物作为磷光发光材料可以发蓝光,并且具有稳定性好,效率高,发光区间窄且在长波长蓝光的区间内,完全适合做高质量的oled相关产品中的有机蓝光发光体。
在本发明中,铂配合物用作有机光电装置的蓝磷光发光材料具有能量低、色纯度好的特点,全面超越现在使用的荧光材料,将改变目前平板显示中缺少稳定高效蓝光掺杂材料的情况,进发射光色并改善器件的性能。更明确地说,此类稳定的配合物发光材料的cie坐标和发光效率将被调谐成更符合平板显示的需求。
本发明的目的之三在于提供一种有机光电装置,所述有机光电装置包括有机层,所述有机层包括发光层以及功能层;
在本发明中,功能层是指除发光层之外的有机光电装置中的其它层,示例性地如电子传输层、空穴注入层、空穴传输层等。
优选地,所述发光层包括如目的之一所述的化合物中的任意一种或至少两种的组合;
优选地,所述功能层包括如目的之一所述的化合物中的任意一种或至少两种的组合。
为达到此发明目的,本发明采用以下技术方案:
相对于现有技术,本发明具有以下有益效果:
本发明提供了一种新型的铂配合物,通过在甲基吡啶并咪唑型卡宾引入二价铂配合物配体,使得到的配合物具有较高的稳定性和发光效率,发光区间窄且在长波长蓝光区间内,能够作为蓝磷光发光材料应用于有机光电装置中;且该铂配合物用作蓝磷光发光材料具有效率高、能量低、色度好等优点,且能够达到发射蓝光光色并改善器件类型。
附图说明
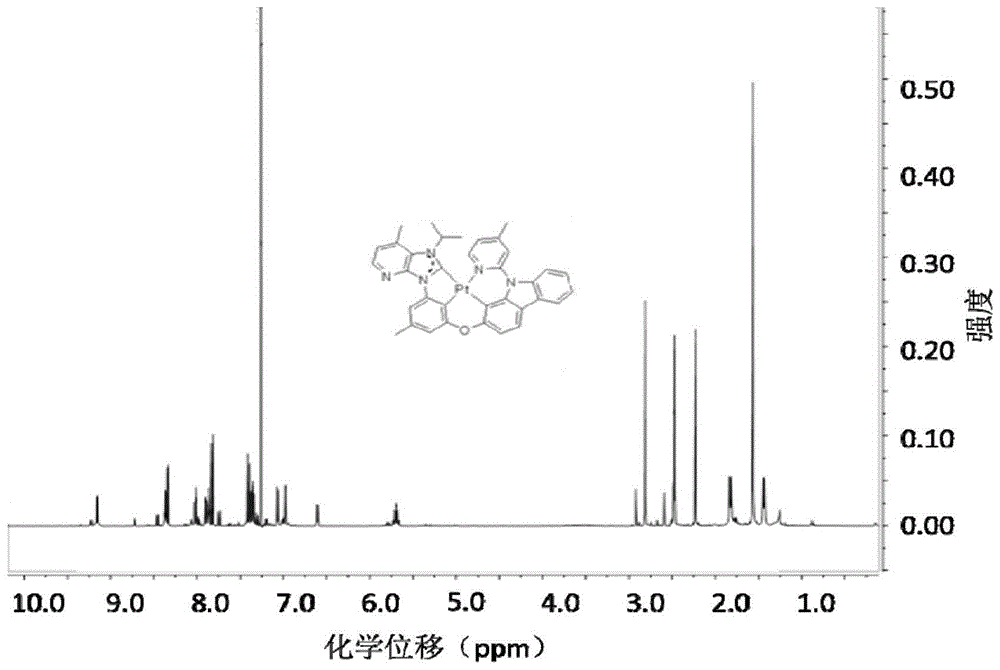
图1是合成例1中得到的铂配合物226的核磁图谱;
图2是合成例1中得到的铂配合物226的质谱图;
图3是合成例2中得到的铂配合物16的液相-质谱图;
图4为合成例5中得到的铂配合物51的质谱图;
图5是合成例1中得到的铂配合物226在溶液中和薄膜中的发光光谱图;
图6是合成例3中得到的铂配合物69在溶液中和薄膜中的发光光谱图;
图7是合成例6中得到的铂配合物227在溶液中和薄膜中的发光光谱图;
图8为具体实施方式中oled器件的结构断面图;
图9为具体实施方式中又一oled器件的结构示意图;
图10为合成例9得到的铂配合物制备的oled器件的发光光谱图;
图11为合成例9和合成对比例2的铂配合物制备的oled器件的电流效率对比图;
图12为合成例9和合成对比例2的铂配合物制备的oled器件的光致发光随时间的衰减图;
图13为合成例9和合成对比例2的铂配合物制备的oled器件的功率效率对比图;
图14为合成例9和合成对比例2的铂配合物制备的oled器件的电致发光随时间的衰减图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
本发明式i化合物的代表合成路径如下:
其中,ra、rb、rc、rd、rf和re的取值范围与上述限定的取值范围相同。
本发明中未提到的合成方法的化合物的都是通过商业途径获得的原料产品。本发明中所用溶剂和试剂,例如2-溴咔唑、2-溴-4-甲基吡啶、碘化亚铜、1-甲基咪唑、甲苯、石油醚、乙酸乙酯、无水硫酸钠、3-氨基-5-甲基苯酚、l-脯氨酸、碳酸铯、二甲亚砜等化学试剂,均可以从国内化工产品市场购买,例如购买自阿拉丁、西格玛、国药集团试剂公司、tci公司、上海毕得医药公司、百灵威试剂公司等。另外,本领域技术人员也可以通过公知方法合成。
具体实施方式中,在cdcl3或dmso-d6溶液中,通过varian液态核磁共振仪记录1hnmr(氢核磁共振)和13cnmr(碳核磁共振),光谱为300、400或500mhz,而化学位移是以残余的质子化溶剂为基准。如果使用cdcl3作为溶剂,则采用四甲基硅烷(δ=0.00ppm)作为内参比来记录1hnmr(氢核磁共振)光谱;采用cdcl3(δ=77.00ppm)作为内参比来记录13cnmr(碳核磁共振)光谱。如果使用dmso-d6作为溶剂,则采用残余的h2o(δ=3.33ppm)作为内参比来记录1hnmr(氢核磁共振)光谱;采用dmso-d6(δ=39.52ppm)作为内参比来记录13cnmr(碳核磁共振)光谱。以下缩写词是用于代表1hnmr(氢核磁共振)的多样性:s=单线态,d=双线态,t=三线态,q=四线态,p=五线态,m=多线态,br=宽。
具体实施方式中cat.指代的意义为催化剂;r.t.指代的意义为室温。
合成例1
铂配合物226的合成
(1)1-1的合成:
制备过程:向带有磁力转子的48ml封管中依次添加2-溴咔唑(3.69g,15mmol),2-溴-4-甲基吡啶0002(1.42ml,15mmol)、碘化亚铜(0.3mmol,0.02equiv)、1-甲基咪唑(0.3mmol,0.2equiv)、t-buoli(18mmol,1.2equiv)和甲苯(50ml),得到的混合物经氮气鼓泡10分钟后加热到120℃搅拌8小时。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。减压蒸馏除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,洗脱剂为石油醚:乙酸乙酯=25:1,得到白色固体,收率93%。
(2)1-2的合成:
制备过程:向25ml的史莱克管中加入中间体1-2(1equiv)、3-氨基-5-甲基苯酚(1.2equiv)、碘化亚铜(10%)、l-脯氨酸(l-pro,20%)、碳酸铯(2equiv)和二甲亚砜(0.5m)。得到的混合物用氮气鼓泡10分钟,并在120℃下搅拌3天。在冷却后,添加水和乙酸乙酯(ea),并将混合物过滤。采用乙酸乙酯提取水相,并将各有机相合并,用卤水清洗,有机相用无水na2so4干燥。采用pe∶ea=8∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,得到产物1-2(褐色粘稠液体,收率75%)。
(3)1-3的合成:
制备过程:将15ml丙酮和25ml乙酸加入8g2-氯-3-氨基-4-甲基吡啶在20ml二氯甲烷的溶液中。在0℃下加入6ml硼烷二甲硫醚溶液,接着室温(r.t.)搅拌过夜。反应完全后加入25wt%氨水溶液调节ph到8。加入50ml水后,用二氯甲烷萃取三次。收集有机相并用无水硫酸钠干燥,旋干得到产物1-3的粗产物直接用于下一步(黄色油状物)。
(4)1-4的合成:
制备过程:向手套箱中的一个密封管添加中间体1-3(1equiv)、中间体1-2(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物1-4(黄色粘稠液,收率85%)。
(4)1-5的合成:
制备过程:向一个封管中添加中间体1-4(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物1-5(棕色固体,收率60%)。
(5)1-5的合成:
制备过程:向一个封管中添加中间体1-4(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物1-5(棕色固体,收率60%)。
(6)铂配合物226的合成:
制备过程:在封管中加入卡宾六氟磷酸盐1-5(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm∶pe=4∶1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物226(亮黄色粉末,收率35%)。
结构表征:
图1为铂配合物226的1hnmr核磁谱图;通过氢谱可以看出该配合物可以独立稳定地存在并且可以分离提纯和表征。从核磁图谱上看,除了该二价铂配合物具有稳定的结构表征之外,二价铂配合物未显示出聚集形态的信号,说明在溶液状态下该类二价铂配合物分子是以单分子分离的状态存在的。
核磁表征的数据为:1hnmr(300mhz,cdcl3)δ9.15–9.16(d,1h),8.36-8.37(m,1h),8.34–8.33(d,1h),8.01-8.03(m,1h),7.91(m,1h),7.86(m,1h),7.81(d,1h),7.39-7.41(d,1h),7.37–7.34(m,2h),7.08–7.06(m,1h),6.98-6.97(m,1h),6.61-6.60(m,1h),5.73-5.66(m,1h),2.81(s,3h),2.48(s,3h),2.23(s,3h),1.82-1.84(d,3h),1.43-1.45(d,3h)。
图2为铂配合物226的质谱图,从图2可知,质谱分子显示分子信号显示m/c峰值为732.5,与配合物226的分子离子峰一致,说明该配合物结构为设计结构。
质谱表征的数据为:ms(esi):732.5[m+1]+.二氯甲烷溶液中发光峰值461nm,半峰宽=47nm,pmma薄膜中发光峰峰值458nm,半峰宽=44nm。
合成例2
铂配合物16的合成
(1)2-1的合成:
制备过程:向带有磁力转子的48ml封管中依次添加2-溴咔唑(3.69g,15mmol),2-溴吡啶0002(1.42ml,15mmol)、碘化亚铜(0.3mmol,0.02equiv)、1-甲基咪唑(0.3mmol,0.2equiv)、t-buoli(18mmol,1.2equiv)和甲苯(50ml),得到的混合物经氮气鼓泡10分钟后加热到120℃搅拌8小时。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。减压蒸馏除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,洗脱剂为石油醚∶乙酸乙酯=25∶1,得到产物2-1,收率95%。
(2)2-2的合成:
制备过程:向25ml的史莱克管中加入中间体2-1(1equiv)、3-氨基-苯酚(1.2equiv)、碘化亚铜(10%)、l-脯氨酸(l-pro,20%)、碳酸铯(2equiv)和二甲亚砜(0.5m)。得到的混合物用氮气鼓泡10分钟,并在120℃下搅拌3天。在冷却后,添加水和乙酸乙酯(ea),并将混合物过滤。采用乙酸乙酯提取水相,并将各有机相合并,用卤水清洗,有机相用无水na2so4干燥。采用pe∶ea=8∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,得到产物2-2(褐色粘稠液体,收率70%)。
(3)2-3的合成:
制备过程:向手套箱中的一个密封管添加中间体1-3(1equiv)、中间体2-2(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物2-3(黄色粘稠液,收率85%)。
(4)2-4的合成:
制备过程:向一个封管中添加中间体2-3(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物2-4(棕色固体,收率50%)。
(4)铂配合物16的合成:
制备过程:在封管中加入卡宾六氟磷酸盐2-4(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm∶pe=4∶1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物26(亮黄色粉末,收率40%)。
图3为配合物16的质谱图,质谱分子显示分子信号显示m/c峰值为704.4,与化合物26的分子离子峰一致,说明该配合物结构为设计结构。
结构表征:ms(esi):704.45[m+h+]二氯甲烷中发光峰值458nm,半峰宽=42nm,pmma中发光峰值457nm,半峰宽=45nm。
合成例3
铂配合物63的合成
(1)3-1的合成:
制备过程:向25ml的史莱克管中加入中间体1-1(1equiv)、3-氨基-苯酚(1.2equiv)、碘化亚铜(10%)、l-脯氨酸(l-pro,20%)、碳酸铯(2equiv)和二甲亚砜(0.5m)。得到的混合物用氮气鼓泡10分钟,并在120℃下搅拌3天。在冷却后,添加水和乙酸乙酯(ea),并将混合物过滤。采用乙酸乙酯提取水相,并将各有机相合并,用卤水清洗,有机相用无水na2so4干燥。采用pe∶ea=8∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,得到产物3-1(褐色粘稠液体,收率69%)。
(2)3-2的合成:
制备过程:将15ml丙酮和25ml乙酸加入8g2-氯-3-氨基-4,6-二甲基吡啶在20ml二氯甲烷的溶液中。在0℃下加入6ml硼烷二甲硫醚溶液,接着室温搅拌过夜。反应完全后加入25wt%氨水溶液调节ph到8。加入50ml水后,用二氯甲烷萃取三次。收集有机相并用无水硫酸钠干燥,旋干得到产物3-2的粗产物直接用于下一步(黄色油状物)。
(3)3-3的合成:
制备过程:向手套箱中的一个密封管添加中间体3-2(1equiv)、中间体3-1(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物3-3(黄色粘稠液,收率82%)。
(4)3-4的合成:
制备过程:向一个封管中添加中间体3-3(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物3-4(棕色固体,收率64%)。
(5)铂配合物63的合成
制备过程:在封管中加入卡宾六氟磷酸盐3-4(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm∶pe=4∶1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物78(亮黄色粉末,收率44%)。
结构表征:ms(esi):731.3[m+1]+.在二氯甲烷中发光峰值为455nm,半峰宽=37nm,pmma薄膜中发光峰值为451nm,半峰宽=23nm。
通过fluorolog-3光量子效率平台测试,室温下薄膜中光量子产率:49%;室温下薄膜中激子寿命:2.2μs。
合成例4
铂配合物69的合成
(1)4-1的合成
制备过程:将15ml丙酮和25ml乙酸加入8g2-氯-3-氨基-4-甲基-6-溴吡啶在20ml二氯甲烷的溶液中。在0℃下加入6ml硼烷二甲硫醚溶液,接着室温搅拌过夜。反应完全后加入25wt%氨水溶液调节ph到8。加入50ml水后,用二氯甲烷萃取三次。收集有机相并用无水硫酸钠干燥,旋干得到产物3-2的粗产物直接用于下一步(黄色油状物)。
(2)4-2的合成:
制备过程:选取100ml圆底烧瓶,加入底物4-1(1eq.)、四(三苯基膦)钯(5%)和溶剂乙二醇二甲醚比水2:1(0.1m),室温搅拌二十分钟后,加入2,4,6-三甲基苯硼酸(1.1eq.)和碳酸钠(3eq.),加热至90摄氏度回流36h。冷却至室温,用乙酸乙酯萃取3次,收集有机相,用无水硫酸钠干燥,旋干后柱层析,石油醚比乙酸乙酯40:1作为洗脱液,得到淡黄色固体,产率60%。
(3)4-3的合成:
制备过程:向手套箱中的一个密封管添加中间体4-2(1equiv)、中间体3-1(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物4-3(黄色粘稠液,收率75%)。
(4)4-4的合成:
制备过程:向一个封管中添加中间体4-3(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物4-4(黄色固体,收率54%)。
(5)铂配合物69的合成
制备过程:在封管中加入卡宾六氟磷酸盐4-4(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm:pe=2:1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物84(黄色粉末,收率44%)。
结构表征:ms(esi):836.6[m+1]+.在二氯甲烷中发光峰值454nm,半峰宽=37nm,在pmma中发光峰值452nm,半峰宽=37nm。
合成例5
铂配合物51的合成:
(1)5-2的合成:
制备过程:向手套箱中的一个密封管添加中间体5-1(1equiv)、中间体3-1(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe:ea=6:1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物5-2(黄色粘稠液,收率70%)。
(2)5-3的合成:
制备过程:向一个封管中添加中间体5-2(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物5-3(黄色固体,收率44%)。
(3)铂配合物51的合成:
制备过程:在封管中加入卡宾六氟磷酸盐5-3(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm:pe=2:1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物51(黄色粉末,收率32%)。
图4为铂配合物51的质谱图,质谱分子显示分子信号显示m/c峰值为794.6,与化合物51的分子离子峰一致,说明该配合物结构为设计结构。
结构表征:ms(esi):793.53[m+h+].在二氯甲烷中发光峰值457nm,半峰宽=36nm,在pmma中发光峰值454nm,半峰宽=23nm。
合成例6
铂配合物227的合成
(1)6-1的合成:
制备过程:选取100ml圆底烧瓶,加入底物4-1(1eq.)、四(三苯基膦)钯(5%)和溶剂乙二醇二甲醚比水2:1(0.1m),室温搅拌二十分钟后,加入2,4,6-三甲基苯硼酸(1.1eq.)和碳酸钠(3eq.),加热至90摄氏度回流36h。冷却至室温,用乙酸乙酯萃取3次,收集有机相,用无水硫酸钠干燥,旋干后柱层析,石油醚比乙酸乙酯40:1作为洗脱液,得到淡黄色固体,产率86%。
(2)6-2的合成:
制备过程:选取合适的封管,加入中间体6-1(1eq.),加入三苯基膦(2eq.),邻二氯苯作为溶剂(0.2m),180℃反应24小时。旋干后硅胶柱层析,石油醚比乙酸乙酯5:1作洗脱剂,得到褐色固体6-2,产率78%。
(3)6-3的合成:
制备过程:向带有磁力转子的48ml封管中依次添加中间体6-2(3.72g,15mmol),2-溴-4甲基吡啶(1.5ml,15mmol)、碘化亚铜(0.3mmol,0.02equiv)、1-甲基咪唑(0.3mmol,0.2equiv)、t-buoli(18mmol,1.2equiv)和甲苯(50ml),得到的混合物经氮气鼓泡10分钟后加热到120℃搅拌8小时。冷却至室温,加水淬灭反应,用乙酸乙酯萃取,合并有机相,用适量饱和氯化钠水溶液洗涤后加无水硫酸钠干燥。减压蒸馏除去溶剂,将所得粗产品通过硅胶柱色谱分离纯化,洗脱剂为石油醚:乙酸乙酯=25:1,得到产物6-3,收率95%。
(4)6-4的合成:
制备过程:选取圆底烧瓶一个,加入合适大小的磁子。将中间体6-3(1eq.)加入烧瓶中,并量取二氯甲烷(0.1m)加入烧瓶中,将反应瓶放置于冰浴中搅拌,接着缓慢滴加三溴化硼(2.5eq.),加完之后室温下搅拌18h.。缓慢加入饱和碳酸氢钠水溶液淬灭,采用二氯甲烷提取水相,并将各有机相合并,用水清洗,有机相用无水na2so4干燥。采用pe∶ea=4:1作为洗脱液,通过硅胶层析来净化所获得的溶液,得到目标产物6-4(灰色固体,收率80%)。
(5)6-5的合成:
制备过程:向25ml的史莱克管中加入中间体6-4(1.1equiv)、3-溴苯胺(1equiv)、碘化亚铜(10%)、l-脯氨酸(l-pro,20%)、碳酸铯(2equiv)和二甲亚砜(0.5m)。得到的混合物用氮气鼓泡10分钟,并在120℃下搅拌3天。在冷却后,添加水和乙酸乙酯(ea),并将混合物过滤。采用乙酸乙酯提取水相,并将各有机相合并,用卤水清洗,有机相用无水na2so4干燥。采用pe∶ea=8∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,得到产物6-5(褐色粘稠液体,收率64%)。
(6)6-6的合成:
制备过程:向手套箱中的一个密封管添加中间体1-3(1equiv)、中间体6-5(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物6-6(黄色粘稠液,收率81%)。
(7)6-7的合成:
制备过程:向一个封管中添加中间体6-6(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物6-7(棕色固体,收率50%)。
(8)铂配合物227的合成:
制备过程:在封管中加入卡宾六氟磷酸盐6-7(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm∶pe=4∶1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物242(亮黄色粉末,收率45%)。
结构表征:ms(esi):718.3[m+h+].在二氯甲烷中发光峰值457nm,半峰宽=41nm,在pmma中发光峰454nm,半峰宽=40nm。
常温下薄膜中量子产率:41%;常温下薄膜中激子寿命:1.5μs。
合成例7
铂配合物228的合成
(1)7-1的合成:
制备过程:向25ml的史莱克管中加入中间体6-4(1.1equiv)、2-甲基-3-溴苯胺(1equiv)、碘化亚铜(10%)、l-脯氨酸(l-pro,20%)、碳酸铯(2equiv)和二甲亚砜(0.5m)。得到的混合物用氮气鼓泡10分钟,并在120℃下搅拌3天。在冷却后,添加水和乙酸乙酯(ea),并将混合物过滤。采用乙酸乙酯提取水相,并将各有机相合并,用卤水清洗,有机相用无水na2so4干燥。采用pe∶ea=8∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,得到产物7-1(褐色粘稠液体,收率64%)。
(2)7-2的合成:
制备过程:向手套箱中的一个密封管添加中间体1-3(1equiv)、中间体7-1(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物7-2(黄色粘稠液,收率81%)。
(3)7-3的合成:
制备过程:向一个封管中添加中间体7-2(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物7-3(棕色固体,收率56%)。
(4)铂配合物228的合成:
制备过程:在封管中加入卡宾六氟磷酸盐7-3(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm∶pe=4∶1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物243(亮黄色粉末,收率40%)。
结构表征:ms(esi):732.4[m+h+].在二氯甲烷中发光峰461nm,半峰宽=46nm,在pmma中发光峰在459nm,半峰宽=43nm。
常温下薄膜中量子产率:50%;常温下薄膜中激子寿命:1.4μs。
合成例8
铂配合物21的合成:
(1)8-1的合成:
制备过程:向手套箱中的一个密封管添加中间体1-3(1equiv)、中间体3-1(1.1equiv)、三(二亚苄基丙酮)二钯(5%)、1,1'-联萘-2,2'-双二苯膦(5%)、叔丁醇钠(1.5equiv)和甲苯(0.2m)。在让混合物鼓泡15分钟后,在130℃下将混合物加热20小时。在冷却后,加入乙酸乙酯,然后将混合物过滤。采用乙酸乙酯提取水相,并将各有机相混合,用卤水清洗,用无水na2so4干燥。采用pe∶ea=6∶1作为洗脱液,通过硅胶层析来净化所获得的溶液,将洗脱液旋干,得到产物8-1(黄色粘稠液,收率88%)。
(2)8-2的合成:
制备过程:向一个封管中添加中间体8-1(1equiv)、六氟磷酸铵(1.1equiv)和原甲酸三乙酯(0.5m)。120℃加热过夜。冷却至室温后加入乙酸乙酯析出黄色沉淀,过滤得到产物8-2(棕色固体,收率64%)。
(3)铂配合物21的合成:
制备过程:在封管中加入卡宾六氟磷酸盐8-2(1equiv)、二氯(1,5-环辛二烯)铂(ii)(pt(cod)cl2,0.9equiv)、醋酸钠(1.05equiv)和thf(0.5m)。在120℃下加热3天。冷却至室温后旋干,采用dcm∶pe=4∶1作为洗脱液,通过硅胶层析来纯化所获得的溶液,得到目标产物:铂配合物21(亮黄色粉末,收率33%)。
结构表征:ms(esi):718.2[m+h+].在二氯甲烷中发光峰在461nm,半峰宽=46nm,在pmma中发光峰在459nm,半峰宽=43nm。
合成例9
铂配合物47的合成
(1)9-3的合成:
制备过程:与合成例2的区别仅在于将2-3合成过程中采用的中间体1-3替换为9-2,其余制备过程均与2-3的合成过程相同;得到产物9-3(黄色泡沫物,收率86%)。
(2)9-4的合成:
制备过程:与8-2的合成过程的区别仅在于将8-2制备过程中采用的8-1替换为9-3,其余合成过程均与8-2的合成过程相同;得到产物9-4(棕色固体,收率70%)。
(3)铂配合物47的合成:
制备过程:与合成例8中的铂配合物21的合成步骤的区别仅在于将铂配合物21合成过程中采用的8-2替换为9-4,其余合成过程均相同;得到目标产物:铂配合物47(亮黄色粉末,收率33%)。
结构表征:ms(esi):878.3[m+1]+.在二氯甲烷中发光峰在455nm,半峰宽=26nm,在pmma中发光峰在458nm,半峰宽=25nm。
实施例1
本实施方式的合成例的铂配合物的发射光谱显示,在向配体添加氘原子,三线态能隙几乎是相同的。
本实施方式中合成例得到的铂配合物的蓝光波长峰值在450-470区间内,更进一步讲蓝光的光谱超过70%在450-490nm区间内。
发射体色纯度的代表性数据从其采用5%pmma(聚甲基丙烯酸甲酯)三氯甲烷溶液制备的薄膜发射光谱获得。
图5为铂配合物226在溶液中和薄膜中的发光光谱图;从图5可以看出,薄膜发射峰值波长在458nm,较溶液发射波长461nm有蓝移效应,两者都显示出较好的蓝光发光光谱,说明该配合物226适合做蓝光发光方面的应用。
图6为铂配合物69在溶液中和薄膜中的发光光谱图,从图6可以看出,薄膜发射峰值波长在452nm,较溶液发射波长454nm有蓝移效应,两者都显示出较好的蓝光发光光谱,说明该配合物69适合做蓝光发光方面的应用。
图7为铂配合物227在溶液中和薄膜中的发光光谱图,从图7可以看出,薄膜发射峰值波长在454nm,较溶液发射波长457nm有蓝移效应,两者都显示出较好的蓝光发光光谱,说明该配合物227适合做蓝光发光方面的应用。
表1中,λ为峰值波长,fwhm为半峰宽,450-700nm为发射光谱在450-500nm区间内的积分的比例,cie(x,y)是根据国际照明委员会标准的色度坐标参数。配合物226、16、63、69、51、227、228、21、47的峰值波长在455-459之间,半峰宽在23-44nm之间。在符合视觉健康的蓝光发光区域450-500nm区间范围之内的光谱占比达到60%以上。并且,铂配合物都在深蓝光发光区间之内(x<0.15,y<0.17),低能量、窄光谱的特性保证了这类二价铂配合物成为极其优秀的蓝光材料。其中配合物63较配合物r1、配合物47较配合物r2在光谱更窄、发光效率提高360%和60%,因此铂配合物63和47更适合做纯蓝光的发射体。对比配合物r3、r4,项比较与本专利的合成例化合物有明显的红移,并且半峰宽变宽,已经不适合做纯蓝光器件,其中r3为天蓝光,r4为蓝绿光。
表1
配合物226、16、63、69、51、227、228、21、47的带隙和光学性质表征如下:材料的带隙值(eg)、lumo和homo值采用循环伏安法(cv)测得。整个测试过程在手套箱(lab2000,etelux)中的chi600d电化学工作站(上海辰华仪器公司)上进行,以pt柱为工作电极、以ag/agcl为参比电极,pt丝为辅助电极构成三电极系统,测试过程采用的介质是0.1m六氟磷酸四丁基胺(bu4npf6)的二甲基甲酰胺(dmf)溶液,所测电势均以加入的二茂铁(fc)作为内标。下表中λ为二价铂铂配合物溶解在二氯甲烷中的峰值波长,fwhm为其半峰宽,材料的三线态光子能量(et1)由公式1240/λ0→1计算而来(λ0→1为77k条件下的第一振动峰),单位为电子福特(ev)。
表2
本实施方式提供一种oled器件,如图8所示,oled器件1000包括基体1002、阳极层1004、空穴传输层1006、发光层1008、电子传输层1010以及金属阴极层1012。
其中阳极层1004通常为透明材料,示例性地如铟锡氧(ito)、铟锌氧(izo)、二氧化锡(sno2)、氧化锌(zno)等氧化物透明导电材料和它们的任意组合。
空穴传输层1006可以为单层结构的空穴传输层(htl),包括只含有一种化合物的单层空穴传输层和含有多种化合物的单层空穴传输层。空穴传输区也可以为包括空穴注入层(hil)、空穴传输层(htl)、电子阻挡层(ebl)中的至少一层的多层结构;空穴传输区的材料可以选自、但不限于酞菁衍生物如cupc、导电聚合物或含导电掺杂剂的聚合物如聚苯撑乙烯、聚苯胺/十二烷基苯磺酸(pani/dbsa)、聚(3,4-乙撑二氧噻吩)/聚(4-苯乙烯磺酸盐)(pedot/pss)、聚苯胺/樟脑磺酸(pani/csa)、聚苯胺/聚(4-苯乙烯磺酸盐)(pani/pss)、芳香胺衍生物等。
发光层1008可以是包括一发射体和一主体的发光材料,其中发光材料选自合成例制备得到的化合物,也可选择性地连带一种主体材料。主体材料可以是本技术中已知的任何合适的主体材料。oled的发光颜色由发光层1008材料的发光能量(光学能隙)决定,可以按照如上所述通过调谐发射二价铂铂配合物和/或主体材料的电子结构来调谐发光层1008材料的发光能量(光学能隙)。
电子传输层1010可以为单层结构的电子传输层,包括只含有一种化合物的单层电子传输层和含有多种化合物的单层电子传输层。电子传输区也可以为包括空穴阻挡层(ebl)、电子传输层、阴极覆盖层(cpl)中的至少一层的多层结构;空穴传输区的材料可以选自芳香胺、含咔唑的小分子,但不限于上述小分子结构单元。
金属阴极层1012的材料选自镁(mg)、银(ag)、铝(al)、铝-锂(al-li)、钙(ca)、镁-铟(mg-in)、镁-银(mg-ag)等金属或合金以及它们之间的任意组合。
在本实施方式中,oled器件的结构示例性地为:ito/pedot∶pss(70nm)/主体材料∶铂配合物(1000-x∶x,40nm)/dpepo(10nm)/tmpypb(50nm)/liq(1nm)/al(100nm)。
本实施方式中,空穴传输层1006、发光层1008、电子传输层1010可以通过真空热蒸镀、旋转涂敷、打印等方法形成于电极之上。用作有机材料层的化合物可以为有机小分子、有机大分子和聚合物,以及它们的组合。
本实施方式中,oled主要通过常规的旋涂法制备,示例性的制备过程包括:将ito基板在紫外臭氧环境中处理20min,然后使用匀胶机制备pedot:pss(4000rpm,40s)层,旋涂制备铂配合物与主体材料发光层(6000rpm,60s),使用真空蒸镀设备在真空环境下(<5.0*10^-4pa)以0.1a/s蒸镀制备dpepo层及tmpypb层,使用真空蒸镀设备以速率为0.1a/s和0.5a/s分别制备lif层及al电极层。
实施例2
将合成例1-9和对比合成例1-4得到的铂配合物作为发光材料掺杂到主体材料中制备成oled器件,其中oled器件的结构示意图如图9所示,包括阳极层12、空穴注入层14、空穴传输层16、电子阻挡层18、发光层20、空穴阻挡层22、电子传输层24和金属阴极层26。
本实施例中oled器件的结构为:ito/p-dopinght/htl/ebl/主体材料∶铂配合物/hbl/n-dopinget/金属电极,p-doping和n-doping分别指掺杂p型和n型材料,其材料可以是有机材料也可以是无机材料。具体选自:ito/hatcn(10nm)/npd(40nm)/tapc(10nm)/95%2,6-mcpy:5%配合物47(25nm)/2,6-mcpy(10nm)/tmpypb(30nm)/lif(1nm)/al(100nm)
图10为铂配合物47制备得到的oled器件的电致发光光谱图,根据图10可知,发光峰相对其在pmma介质中的光致发光峰红移5nm,半峰宽相当,保持了发光配合物47本身的发光特性,计算得到其色度坐标值为cie(0.15,0.10),说明此器件适合用作深蓝光发光器件。根据归一化积分显示,小于450nm的刺激性蓝光的成分只有14.5%、70.2%的光子能量在500nm以上。根据传统的蓝光归属,在450-500nm之间的蓝光光子占所有发射光子的数的55.7%。
图11为铂配合物47和r2所制备的器件的光转化电流效率图,由图11可以看出,使用配合物47制备的器件电流效率更高。图11显示了使用配合物47器件光电转换电流效率曾现非常稳定的曲线,从0ma/cm2到20ma/cm2电流密度变化下,电流滚降小于10%,并且该掺杂材料的器件具有较高的效率,其中2%和5%掺杂的器件在10ma/cm2电流效率分别为13.4和15.4cd/a,说明配合物47作为蓝光发光掺杂材料具有高效的、稳定发光的光转化性能。
图12为铂配合物47和r2所制备的器件的光致发光随时间的衰减曲线图,由图12可以看出,使用配合物47和r25wt%的掺杂的聚苯乙烯(ps)薄膜在375nmuv光照射下发光强度随着时间的变化,说明该器件使用配合物47在强发光条件下60分钟能保持衰减不超过2%,6小时内的光谱衰减小于10%,此实验说明络合物具有极好的发光稳定性。本次衰减测试是通过50mw/m2的紫外光照射掺杂5%的材料的稳定聚苯乙烯高分子薄膜,并记录光致发光的强度,最终得到化合物发光衰减和时间的函数关系。图12显示,配合物47发光强度保持要明显高于对比材料配合物r2,其光致发光衰减的更慢,具有更好的发光稳定性。
图13为铂配合物47和r2所制备的器件光电转换中的功率效率配合物47在发光层材料中占比2%和5%。其中2%掺杂的器件中截取的功率效率在1147cd/m2亮度下为11lm/w,5%掺杂的器件中截取的功率效率在1341cd/m2亮度下为14.1lm/w,说明配合物47作为蓝光发光掺杂材料具有高效的、稳定发光的光转化性能。通过图13所示配合物47与对比材料r2制备的器件的光电转化功率效率对比图可以看出,使用配合物47制备的器件功率效率更高,说明引入甲基可以提升器件功效。
图14为铂配合物47和r2所制备的器件光致发光随时间的衰减曲线,由图14可知,配合物47较r2掺杂的器件电致发光衰减的较慢,具有更好的器件稳定性。lt95@20ma/cm2的寿命分别为78.6分钟和54分钟,使用配合物47掺杂的器件在同等条件下要比r2的器件稳定性高。
将合成例1-9和对比合成例1-4得到的铂配合物作为发光材料掺杂到主体材料中制备成oled器件中,对其进行性能测试,测试结果见表3:
表3
如表3所示为各铂铂配合物制备的发光器件的发光性能数据对比。通过铂配合物r1较63和r2较47对比发现,含有4-吡啶甲基的铂配合物63和47明显具有更好的器件应用性能,其发光效率提升明显;其中铂配合物发光器件的电致发光波长主要由铂铂配合物本身光致发光的决定,铂铂配合物本身光致发光光谱的纯度和电致发光的光谱纯度直接相关。在同一条件下,发光器件的效率高低也与铂配合物本身的发光量子效率趋势一致,发光器件的发光的色纯度与掺杂材料本身光激发下发射光的光谱色纯度直接关联。铂铂配合物发光器件的电致发光光谱和薄膜中光致发光器件比较可知,相比薄膜光致发光光谱,发光器件的电致发光光谱稍有红移,但波峰波长仍位于蓝光区域(460-470nm),光谱大部分也位于蓝光范围内,计算的色度坐标说明该发光器件属于纯蓝光发光器件。由于大部分光都在蓝光区间,仅仅需要滤除少量长波长的光,说明本发明的实施方式所提供的络合物材料就可以完全满足显示器中的高效率纯蓝光cie(0.14,0.08)器件的色度要求。
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!