一种新型非金属催化剂催化酮和α,β-不饱和酮的不对称氢化
一种新型非金属催化剂催化酮和
α
,
β-不饱和酮的不对称氢化
技术领域
1.本发明涉及一种新型非金属催化剂催化酮和α,β-不饱和酮的不对称氢化,属于有机合成技术领域。
背景技术:
2.醇类和四氢萘酮类化合物是许多具有生物活性分子的核心骨架,具备一定的抗菌、抗癌活性。目前,制备此类化合物往往需要手性金属催化剂参与,面临着金属残余污染等一系列问题。因此需要提供新型的催化剂。
技术实现要素:
3.本发明的目的是提供一种新型非金属催化剂催化酮和α,β-不饱和酮的不对称氢化,采用手性噁唑啉作碱配合非手性硼烷构成受阻lewis酸碱对(简称flps),简单酮类或α,β不饱和酮类化合物作为原料,以成本低廉的氢气作为氢源,高效、高对映选择性地得到一系列三级醇类或四氢萘酮类化合物,具有潜在的应用价值。
4.本发明所提供的手性噁唑啉化合物的结构式如式ⅰ所示:
[0005][0006]
式i中,r1为碳原子数为6~7的芳基;r2为h或碳原子数为1~4的烷基或碳原子数为6~7的芳基;r3为碳原子数为1~6的烷基或碳原子数为6~7的芳基;r4和r5独立地选自碳原子数为1~6的烷基。
[0007]
具体地,r1优选为苯基或取代苯基,如4-溴苯基;
[0008]
r2优选为h、碳原子数为1~3的烷基或苯基;
[0009]
r3优选为碳原子数为1~3的烷基或苯基;
[0010]
r4优选为碳原子数为1~3的烷基;
[0011]
r5优选为碳原子数为1~3的烷基。
[0012]
本发明提供的手性噁唑啉化合物具体可如下述任一种:
[0013][0014]
本发明所提供的式i所示的噁唑啉催化剂可按照包括下述步骤的方法制备得到的:
[0015]
1)在碱的存在下,式ii所示化合物与式iii所示化合物进行反应,得到对应的酰胺化合物;
[0016]
2)再在钼氨酸(nh4)6mo7o
24
·
4h2o的催化下反应,得到式i所示手性噁唑啉化合物;
[0017][0018]
式ii和式iii中,r
1-r5的定义同式ⅰ。
[0019]
上述方法中,步骤1)中,式ii所示化合物、式iii所示化合物与所述碱的摩尔比可为1:1:1.5。
[0020]
所述反应在两口圆底烧瓶中进行,用氮气保护。
[0021]
所述反应在有机溶剂中进行,所述有机溶剂具体可为二氯甲烷。
[0022]
所述反应温度可为室温,时间为18~24小时。
[0023]
为了保证反应体系的均一性,所述反应在搅拌下进行。
[0024]
上述方法中,步骤2)中,所述酰胺化合物与所述钼氨酸的摩尔比为1:0.25。
[0025]
所述反应在两口圆底烧瓶中进行,安装分水器和回流冷凝管,用氮气保护。
[0026]
所述反应在有机溶剂中进行,所述有机溶剂具体可为对二甲苯。
[0027]
所述反应温度可为145℃,时间为24小时。
[0028]
为了保证反应体系的均一性,所述反应在搅拌下进行。
[0029]
上述方法还进一步包括通过柱层析分离得到式i所示手性噁唑啉化合物的步骤。
[0030]
所述柱层析中,所用层析柱的规格为30mm
×
300mm(直径
×
高度);所用柱填料为硅胶(200~300目);洗脱剂为石油醚与乙酸乙酯的混合溶液(体积比20:1)。
[0031]
本发明所述手性噁唑啉化合物与三(4-氢四氟苯基)硼配合成受阻lewis酸碱对,能够不对称催化氢化酮类化合物制备醇类化合物,具体的步骤如下:
[0032]
在三(4-氢四氟苯基)硼和所述手性噁唑啉化合物的催化下,式v所示酮类化合物与氢气进行反应,得到式iv所示手性醇类化合物;
[0033][0034]
r6为碳原子数为1~4的烷基或碳原子数为6~9的芳基;r7为碳原子数为1~4的烷基。
[0035]
具体地,r6优选为苯基、苄基或取代苯基,所述取代苯基中的取代基可为氟、氯、溴、三氟甲基、-coor1(r1为碳原子数为1~3的烷基)、-cor2(r2为碳原子数为1~3 的烷基);
[0036]
r7优选为碳原子数为1~3的烷基。
[0037]
本发明制备得到了如下醇类化合物:
[0038][0039]
上述的制备方法中,所述三(4-氢四氟苯基)硼、所述手性噁唑啉化合物与式v 所示化合物的摩尔比为1:1~2:10;
[0040]
所述氢气的压力为40~50bar。
[0041]
上述的制备方法中,所述反应在有机溶剂中进行;所述有机溶剂为甲苯和环己烷的混合液。
[0042]
上述的制备方法中,所述反应的温度为30℃~40℃,时间为24~72小时。
[0043]
上述方法还进一步包括通过柱层析分离得到式iv所示醇类化合物的步骤:
[0044]
所述柱层析中,所用层析柱的规格为12mm
×
160mm(直径
×
高度);所用柱填料为硅
胶(200~300目);洗脱剂为正戊烷与n,n
’-
二甲基乙基胺的混合溶液(体积比为 150:1),增加极性正戊烷与乙醚的混合溶液(体积比4:1)。
[0045]
本发明所述手性噁唑啉化合物与三(4-氢四氟苯基)硼配合成受阻lewis酸碱对,能够不对称催化氢化α,β-不饱和酮类化合物制备手性四氢萘酮类化合物,具体的步骤如下:
[0046]
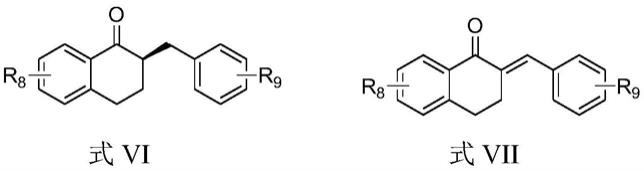
在三(4-氢四氟苯基)硼和权利要求1所述手性噁唑啉化合物的催化下,式vii 所示α,β-不饱和酮类化合物与氢气进行反应,得到式vi所示手性四氢萘酮类化合物;
[0047][0048]
r8为h、氯、溴、碳原子数为1~4的烷基或碳原子数为6~8的芳基;
[0049]
r9为h、氟、氯、溴、碳原子数为1~4的烷基、碳原子数为1~4的烷氧基或-coor, r为碳原子数为1~4的烷基。
[0050]
具体地,r8优选为h、溴、碳原子数为1~3的烷基或苯基;
[0051]
r9优选为h、氯、碳原子数为1~3的烷基、碳原子数为1~3的烷氧基或-coome。
[0052]
本发明制备得到了如下手性四氢萘酮类化合物:
[0053][0054][0055]
上述的制备方法中,所述三(4-氢四氟苯基)硼、所述手性噁唑啉化合物与式vii 所示α,β-不饱和酮类化合物的摩尔比为1:1~2:10;
[0056]
所述氢气的压力为40~50bar。
[0057]
上述的制备方法中,所述反应在有机溶剂中进行;所述有机溶剂为均三甲苯。
[0058]
上述的制备方法中,其特征在于:所述反应的温度为30℃~40℃,时间为24~72 小时。
[0059]
为了保证反应体系的均一性,所述反应在搅拌下进行。
[0060]
上述方法还进一步包括通过柱层析分离得到式vi所示手性四氢萘酮类化合物的步骤;
[0061]
所述柱层析中,所用层析柱的规格为12mm
×
160mm(直径
×
高度);所用柱填料为硅胶(300~400目);洗脱剂为石油醚与二氯甲烷的混合溶液(体积比为10:1)。
[0062]
本发明以多种取代类型的酮类化合物(式v)或α,β-不饱和酮类化合物(式vii) 为原料,在三(4-氢四氟苯基)硼和手性噁唑啉催化下,以氢气为氢源,高效、高立体选择性分别合成了手性醇类化合物(式iv)或手性四氢萘酮类化合物(式vi)。
[0063]
本发明方法具有原料易合成、反应条件温和、操作简便、立体选择性高等优点,产物ee值高达92%,产量高达99%。
具体实施方式
[0064]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0065]
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0066]
实施例1、式i-a所示化合物的制备
[0067]
按照下述合成路线制备式i-a所示化合物:
[0068][0069]
称取上述化合物三乙胺(0.6072g,6mmol)和氨基醇(0.8531g,4mmol)溶于二氯甲烷(4.0ml)中,在冰浴下滴加酰氯(0.4823g,4mmol)的二氯甲烷(4.0ml) 溶液,放入室温油浴中搅拌18h。加入水(50.0ml),乙酸乙酯(3
×
50ml)萃取,合并有机层,用卤水:1m盐酸水溶液=1:1(3
×
50ml)洗,卤水洗(3
×
50ml),无水硫酸钠干燥,旋干,得到白色固体的酰胺。称取上述酰胺化合物(0.4461g,1.5mmol) 和钼酸铵(nh4)6mo7o
24
·
4h2o(0.4634g,0.375mmol)于两口圆底烧瓶中,安装分水器和回流冷凝装置,加入对二甲苯(30.0ml),放入145℃油浴中搅拌24h。反应完后冷至室温,旋干。硅胶柱层析提纯,得到所示白色固体i-a。
[0070]
结构确证结果如下:m.p.61-63℃;[α]
d20
=-171.3(c 0.97,chcl3);ir(film):2971, 1661,1454,1136,978,696cm-1
;1h nmr(300mhz,cdcl3,ppm)δ7.13-6.94(m,6h), 6.94-6.76(m,4h),5.82(d,j=10.2hz,1h),5.51(d,j=10.2hz,1h),1.44(s,9h);
13
c nmr(75mhz,cdcl3,ppm)δ175.4,138.3,137.2,127.9,127.8,127.7,127.4,127.0. 126.5,85.3,74.1,33.9,28.2;hrms(esi)calcd.for c
19
h
22
no(m+h):280.1696,found: 280.1694.
[0071]
经结构鉴定所合成的化合物确为式i-a所示目标化合物(4s,5r)-2-叔丁基-4,5-二苯基噁唑啉。
[0072]
实施例2、i-b所示化合物的制备
[0073]
按照下述合成路线制备式i-b所示化合物:
127.7,127.3,127.1,127.0,126.4,126.2,85.4,74.1,41.2,27.8,27.1;hrms(esi)calcd.for c
20
h
24
no(m+h):342.1852,found:342.1851.
[0083]
经结构鉴定所合成的化合物确为式i-c所示目标化合物(4s,5r)-4,5-二苯基-2-(2-苯基丙-2-基)噁唑啉。
[0084]
实施例4、式i-d所示化合物的制备
[0085]
按照下述合成路线制备式i-d所示化合物:
[0086][0087]
称取上述化合物三乙胺(0.3034g,3mmol)和氨基醇(0.3024g,2mmol)溶于二氯甲烷(4.0ml)中,在冰浴下滴加酰氯(0.2412g,2mmol)的二氯甲烷(4.0ml) 溶液,放入室温油浴中搅拌18h。加入水(50.0ml),乙酸乙酯(3
×
50ml)萃取,合并有机层,用卤水:1m盐酸水溶液=1:1(3
×
50ml)洗,卤水洗(3
×
50ml),无水硫酸钠干燥,旋干,得到白色固体的酰胺。称取上述酰胺化合物(0.3530g,1.5mmol) 和钼酸铵(nh4)6mo7o
24
·
4h2o(0.4634g,0.375mmol)于两口圆底烧瓶中,安装分水器和回流冷凝装置,加入对二甲苯(30.0ml),放入145℃油浴中搅拌24h。反应完后冷至室温,旋干。硅胶柱层析提纯,得到所示无色液体i-d。
[0088]
结构确证结果如下:[α]
d20
=-7.3(c 1.02,chcl3);ir(film):2975,1655,1454,1146, 700cm-1
;1h nmr(300mhz,cdcl3,ppm)δ7.38-7.19(m,3h),7.13(d,j=7.2hz,2h), 5.18(d,j=9.6hz,1h),4.99-4.76(m,1h),1.32(s,9h),0.83(d,j=6.6hz,3h);
13
c nmr (75mhz,cdcl3,ppm)δ175.3,138.5,128.2,127.8,127.4,79.6,71.9,33.5,28.1,16.9; hrms(esi)calcd.for c
14
h
20
no(m+h):218.1539,found:218.1540.
[0089]
经结构鉴定所合成的化合物确为式i-d所示目标化合物(4s,5r)-2-叔丁基-5-甲基-4
-ꢀ
苯基噁唑啉。
[0090]
实施例5、式i-e所示化合物的制备
[0091]
按照下述合成路线制备式i-e所示化合物:
[0092][0093]
称取上述化合物三乙胺(0.4554g,4.5mmol)和氨基醇(0.4115g,3mmol)溶于二氯甲烷(4.0ml)中,在冰浴下滴加酰氯(0.3617g,3mmol)的二氯甲烷(4.0ml) 溶液,放入室温油浴中搅拌18h。加入水(50.0ml),乙酸乙酯(3
×
50ml)萃取,合并有机层,用卤水:1m盐酸水溶液=1:1(3
×
50ml)洗,卤水洗(3
×
50ml),无水硫酸钠干燥,旋干,得到白色固体的酰胺。称取上述酰胺化合物(0.4426g,2mmol) 和钼酸铵(nh4)6mo7o
24
·
4h2o(0.6179g,0.5mmol)于两口圆底烧瓶中,安装分水器和回流冷凝装置,加入对二甲苯(30.0ml),放入145℃油浴中
搅拌24h。反应完后冷至室温,旋干。硅胶柱层析提纯,得到所示无色液体i-e。
[0094]
结构确证结果如下:[α]
d20
=-84.1(c 0.98,chcl3);ir(film):2972,1655,1480,1137, 699cm-1
;1h nmr(500mhz,cdcl3,ppm)δ7.33(t,j=7.5hz,2h),7.29-7.24(m,1h), 7.21(d,j=7.5hz,2h),5.14(dd,j=10.0,7.5hz,1h),4.58(dd,j=10.0,8.5hz,1h), 4.07(t,j=8.0hz,1h),1.31(s,9h);
13
c nmr(125mhz,cdcl3,ppm)δ175.3,138.5, 128.2,127.8,127.4,79.6,71.9,33.5,28.1,16.9;hrms(esi)calcd.for c
13
h
18
no(m+h): 204.1383,found:204.1382.
[0095]
经结构鉴定所合成的化合物确为式i-e所示目标化合物(s)-2-叔丁基-4-苯基噁唑啉。
[0096]
实施例6、式i-f所示化合物的制备
[0097]
按照下述合成路线制备式i-f所示化合物:
[0098][0099]
称取上述化合物三乙胺(0.7589g,7.5mmol)和氨基醇(0.6859g,5mmol)溶于二氯甲烷(5.0ml)中,在冰浴下滴加酰氯(0.7405g,5mmol)的二氯甲烷(5.0ml) 溶液,放入室温油浴中搅拌18h。加入水(80.0ml),乙酸乙酯(3
×
80ml)萃取,合并有机层,用卤水:1m盐酸水溶液=1:1(3
×
80ml)洗,卤水洗(3
×
80ml),无水硫酸钠干燥,旋干,得到白色固体的酰胺。称取上述酰胺化合物(0.7481g,3mmol) 和钼酸铵(nh4)6mo7o
24
·
4h2o(0.9269g,0.75mmol)于两口圆底烧瓶中,安装分水器和回流冷凝装置,加入对二甲苯(60.0ml),放入145℃油浴中搅拌24h。反应完后冷至室温,旋干。硅胶柱层析提纯,得到所示无色液体i-f。
[0100]
结构确证结果如下:[α]
d22
=-74.3(c 0.99,chcl3);ir(film):2958,1653,1455,1137, 699cm-1
;1h nmr(500mhz,cdcl3,ppm)δ7.39-7.30(m,2h),7.29-7.24(m,1h), 7.24-7.19(m,2h),5.15(dd,j=10.0,8.0hz,1h),4.57(dd,j=10.0,8.5hz,1h),4.04(t,j =8.0hz,1h),1.61-1.53(m,2h),1.39-1.30(m,2h),1.28(s,3h),1.27(s,3h),0.93(t,j= 8.0hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ174.7,143.1,128.8,127.6,126.8,75.0, 69.7,43.4,37.0,26.2,26.1,18.3,14.8;hrms(esi)calcd.for c
15
h
22
no(m+h):232.1696, found:232.1697.
[0101]
经结构鉴定所合成的化合物确为式i-f所示目标化合物(s)-2-(2-甲基戊-2-基)-4-苯基噁唑啉。
[0102]
实施例7、式i-g所示化合物的制备
[0103]
按照下述合成路线制备式i-g所示化合物:
1.65(q,j=7.5hz,6h),0.83(t,j=7.5hz,9h);
13
c nmr(125mhz,cdcl3,ppm)δ 173.7,142.1,131.9,128.6,121.4,74.2,69.2,44.2,26.6,8.4;hrms(esi)calcd.for c
16
h
23
nobr(m+h):324.0958,found:324.0963.
[0113]
经结构鉴定所合成的化合物确为式i-h所示目标化合物(s)-2-叔庚基-4-(4-溴苯基) 噁唑啉。
[0114]
实施例9、式iv-a所示化合物的制备
[0115]
按照下述合成路线制备式iv-a所示化合物:
[0116][0117]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-a苯乙酮(0.0601g,0.5mmol),2.5ml甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为54%,ee值68%。
[0118]
结构确证结果如下:[α]
d27
=+40.0(c 0.46,chcl3)(68%ee)[lit.:[α]
d23
=+32.6(c 0.62,chcl3)(98%ee for(r)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ7.42-7.32(m, 4h),7.30-7.23(m,1h),4.95-4.86(m,1h),1.82(br d,j=1.0hz,1h),1.50(d,j=6.5hz, 3h);
13
c nmr(125mhz,cdcl3,ppm)δ146.0,128.7,127.7,125.6,70.6,25.4.
[0119]
经结构鉴定所合成的化合物确为式iv-a所示目标化合物(r)-苯乙醇。
[0120]
实施例10、式iv-b所示化合物的制备
[0121]
按照下述合成路线制备式iv-b所示化合物:
[0122][0123]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-b苯丙酮(0.0671g,0.5mmol),2.5ml甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在40℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为95%,ee值64%。
[0124]
结构确证结果如下:[α]
d26
=-25.7(c 0.74,chcl3)(64%ee)[lit.:[α]
d20
=-19.0(c 1.00,chcl3),(54%ee for(r)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ7.35-7.26(m, 2h),7.26-7.14(m,3h),4.09-3.94(m,1h),2.78(dd,j=13.5,4.5hz,1h),2.68(dd,j= 13.5,8.0hz,1h),1.60(br s,1h),1.24(d,j=6.5hz,3h);
13
c nmr(125mhz,cdcl3, ppm)δ138.7,129.6,128.7,126.7,69.1,46.0,23.0.
[0125]
经结构鉴定所合成的化合物确为式iv-b所示目标化合物(r)-1-苯基-2-丙醇。
[0126]
实施例11、式iv-c所示化合物的制备
[0127]
按照下述合成路线制备式iv-c所示化合物:
[0128][0129]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-c 4-苯基-2-丁酮(0.0741g,0.5mmol),2.5 ml甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气 (40bar),在40℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为96%,ee 值50%。
[0130]
结构确证结果如下:[α]
d26
=-11.6(c 0.69,chcl3)(50%ee)[lit.:[α]
d25
=-14.1(c 0.50,chcl3),(91%ee for(r)-isomer)];1h nmr(400mhz,cdcl3,ppm)δ7.32-7.23(m, 2h),7.23-7.14(m,3h),3.95-3.68(m,1h),2.83-2.53(m,2h),1.89-1.67(m,2h),1.54(br s, 1h),1.23(d,j=6.4hz,3h);
13
c nmr(100mhz,acetone-d6,ppm)δ143.7,129.2,129.1, 126.4,66.9,42.2,32.9,24.1.
[0131]
经结构鉴定所合成的化合物确为式iv-c所示目标化合物(r)-4-苯基-2-丁醇。
[0132]
实施例12、式iv-d所示化合物的制备
[0133]
按照下述合成路线制备式iv-d所示化合物:
[0134][0135]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-d 2-溴苯丙酮(0.1065g,0.5mmol),2.5ml 甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为97%,ee值75%。
[0136]
结构确证结果如下:[α]
d29
=-29.4(c 0.94,chcl3)(75%ee)[lit.:[α]
d20
=+40.3(c 1.00,ch2cl2),(99%ee for(s)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ7.55(d,j= 8.0hz,1h),7.29-7.21(m,2h),7.13-7.06(m,1h),4.20-4.08(m,1h),2.96(dd,j=14.5, 5.0hz,1h),2.84(dd,j=13.5,8.0hz,1h),1.60(br s,1h),1.27(d,j=6.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ138.3,133.2,131.9,128.4,127.6,125.1,67.7,45.8,23.2.
[0137]
经结构鉴定所合成的化合物确为式iv-d所示目标化合物(r)-1-(2-溴苯基)-2-丙醇。
[0138]
实施例13、式iv-e所示化合物的制备
[0139]
按照下述合成路线制备式iv-e所示化合物:
[0140][0141]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-e 2-氯苯丙酮(0.0843g,0.5mmol),2.5ml 甲苯和
2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为95%,ee值75%。
[0142]
结构确证结果如下:[α]
d30
=-31.0(c 0.91,ch2cl2)(75%ee)[lit.:[α]
d21
=-20.0(c 0.011,ch2cl2),(99%ee for(r)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ7.37(dd,j= 7.5,1.5hz,1h),7.29-7.24(m,1h),7.24-7.14(m,2h),4.21-4.07(m,1h),2.96(dd,j=13.5,5.0hz,1h),2.83(dd,j=13.5,8.0hz,1h),1.55(br s,1h),1.27(d,j=6.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ136.6,134.5,131.9,129.9,128.2,127.0,67.7,43.5, 23.2.
[0143]
经结构鉴定所合成的化合物确为式iv-e所示目标化合物(r)-1-(2-氯苯基)-2-丙醇。
[0144]
实施例14、式iv-f所示化合物的制备
[0145]
按照下述合成路线制备式iv-f所示化合物:
[0146][0147]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-f 2-氟苯乙酮(0.0691g,0.5mmol),2.5ml 甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌48小时,旋蒸浓缩,硅胶柱层析提纯,收率为81%,ee值81%。
[0148]
结构确证结果如下:[α]
d27
=+25.7(c 0.79,chcl3)(81%ee)[lit.:[α]
d23
=+41.7(c 1.0, chcl3)(99%ee for(r)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ7.54-7.45(m,1h), 7.28-7.21(m,1h),7.15(t,j=7.5hz,1h),7.07-6.95(m,1h),5.28-5.09(m,1h),1.92(br d, j=4.0hz,1h),1.52(d,j=6.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ160.0(d,j= 243.9hz),132.8(d,j=13.4hz),129.0(d,j=8.3hz),126.8(d,j=4.5hz),124.5(d,j= 3.1hz),115.5(d,j=21.6hz),64.8(d,j=3.0hz),24.2;
19
f nmr(377mhz,cdcl3,ppm) δ-120.1(s,1f).
[0149]
经结构鉴定所合成的化合物确为式iv-f所示目标化合物(r)-2-氟苯乙醇。
[0150]
实施例15、式iv-g所示化合物的制备
[0151]
按照下述合成路线制备式iv-g所示化合物:
[0152][0153]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-g 2-氯苯乙酮(0.0773g,0.5mmol),2.5ml 甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为74%,ee值87%。
[0154]
结构确证结果如下:[α]
d29
=+49.6(c 0.51,chcl3)(87%ee)[lit.:[α]
d23
=+47.19(c 1.53,chcl3)(99%ee for(r)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ7.59(dd,j
=8.0, 1.5hz,1h),7.35-7.27(m,2h),7.20(td,j=8.0,1.5hz,1h),5.37-5.24(m,1h),2.01(br d, j=3.5hz,1h),1.49(d,j=6.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ143.2,131.9, 129.6,128.6,127.4,126.6,67.2,23.7.
[0155]
经结构鉴定所合成的化合物确为式iv-g所示目标化合物(r)-2-氯苯乙醇。
[0156]
实施例16、式iv-h所示化合物的制备
[0157]
按照下述合成路线制备式iv-h所示化合物:
[0158][0159]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-h 2-溴苯乙酮(0.0995g,0.5mmol),2.5ml 甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为43%,ee值86%。
[0160]
结构确证结果如下:[α]
d27
=+46.6(c 0.5,chcl3)(86%ee)[lit.:[α]
d26
=-45.2(c 1.72, chcl3)(99%ee for(s)-isomer)];1h nmr(300mhz,cdcl3,ppm)δ7.60(dd,j=7.8,1.5 hz,1h),7.51(d,j=7.8hz,1h),7.35(t,j=7.5hz,1h),7.13(td,j=8.1,1.8hz,1h), 5.25(q,j=6.3hz,1h),1.98(br s,1h),1.49(d,j=6.3hz,3h);
13
c nmr(125mhz, cdcl3,ppm)δ144.8,132.9,129.0,128.1,126.9,122.0,69.4,23.8.
[0161]
经结构鉴定所合成的化合物确为式iv-h所示目标化合物(r)-2-溴苯乙醇。
[0162]
实施例17、式iv-i所示化合物的制备
[0163]
按照下述合成路线制备式iv-i所示化合物:
[0164][0165]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-i 1-(4-三氟甲基苯基)丙酮(0.1011g,0.5 mmol),2.5ml甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌48小时,旋蒸浓缩,硅胶柱层析提纯,收率为 95%,ee值76%。
[0166]
结构确证结果如下:[α]
d27
=+24.7(c 1.30,chcl3)(76%ee)[lit.:[α]
d22
=-22.0(c 0.50,chcl3),(86%ee for(r)-isomer)];1h nmr(500mhz,cdcl3,ppm):δ7.60(d,j=8.5hz,2h),7.52-7.39(m,2h),4.76-4.62(m,1h),1.91(br s,1h),1.87-1.68(m,2h),0.93 (t,j=7.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ148.7,129.9(q,j=32.0hz), 126.4,125.5(q,j=3.6hz),124.4(q,j=270.3hz),75.5,32.3,10.1;
19
f nmr(377mhz, cdcl3,ppm)δ-62.5(s,3f).
[0167]
经结构鉴定所合成的化合物确为式iv-i所示目标化合物(r)-1-(4-三氟甲基苯基)
ꢀ-
1-丙醇。
[0168]
实施例18、式iv-j所示化合物的制备
[0169]
按照下述合成路线制备式iv-j所示化合物:
[0170][0171]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-j 4-乙酰基苯甲酸甲酯(0.0891g,0.5mmol), 2.5ml甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌48小时,旋蒸浓缩,硅胶柱层析提纯,收率为89%,ee 值74%。
[0172]
结构确证结果如下:[α]
d28
=+30.5(c 0.66,chcl3)(74%ee)[lit.:[α]
d20
=+36.2(c 1.09,chcl3)(96%ee for(r)-isomer)];1h nmr(300mhz,cdcl3,ppm)δ8.00(d,j=8.4 hz,2h),7.43(d,j=8.1hz,2h),4.95(q,j=6.3hz 1h),3.91(s,3h),2.14(br s,1h),1.50 (d,j=6.3hz,3h);
13
c nmr(75mhz,cdcl3,ppm)δ167.2,151.3,129.9,129.0,125.4, 69.9,52.2,25.3.
[0173]
经结构鉴定所合成的化合物确为式iv-j所示目标化合物(r)-4-(1-羟基乙基)苯甲酸甲酯。
[0174]
实施例19、式iv-k所示化合物的制备
[0175]
按照下述合成路线制备式iv-k所示化合物:
[0176][0177]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-k 4-乙酰基苯乙酮(0.0811g,0.5mmol), 2.5ml甲苯和2.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为29%,ee 值74%。
[0178]
结构确证结果如下:[α]
d27
=+41.5(c 0.56,chcl3)(74%ee)[lit.:[α]
d25
=-44.9(c 1.20,chcl3)(98%ee for(s)-isomer)];1h nmr(500mhz,cdcl3,ppm)δ8.01-7.86(m, 2h),7.47(d,j=8.0hz,2h),5.03-4.91(q,j=6.5hz,1h),2.60(s,3h),2.06(br d,j=3.0 hz,1h),1.50(d,j=6.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ198.1,151.4,136.5, 128.8,125.7,70.1,26.8,25.5.
[0179]
经结构鉴定所合成的化合物确为式iv-k所示目标化合物(r)-4-乙酰基苯乙醇。
[0180]
实施例20、式iv-l所示化合物的制备
[0181]
按照下述合成路线制备式iv-l所示化合物,具体操作如下:
[0182][0183]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-a(0.0279g,0.10mmol),式v-l 1,3,5-三乙酰基苯(0.1021g,0.5mmol), 4.5ml甲苯和0.5ml环己烷,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为28%,ee 值78%。
[0184]
结构确证结果如下:[α]
d27
=+23.3(c 0.54,chcl3)(78%ee);ir(film):3421,2972, 1685,1226,894cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.42(t,j=1.5hz,1h), 8.21-8.11(m,2h),5.12-4.96(m,1h),2.67(s,6h),2.07(br d,j=3.5hz,1h),1.56(d,j= 6.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ197.6,147.4,137.9,129.7,127.4,69.8, 27.0,25.8;hrms(apci)calcd.for c
12
h
13
o2(m+h
+-h2o):189.0910,found:189.0907.
[0185]
经结构鉴定所合成的化合物确为式iv-l所示目标化合物(r)-3,5-二乙酰基苯乙醇。
[0186]
实施例21、式vi-a所示化合物的制备
[0187]
按照下述合成路线制备式vi-a所示化合物:
[0188][0189]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-a(e)-2-苯亚甲基-1-四氢萘酮(0.1172g,0.5 mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40 bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为94%,ee值77%。
[0190]
结构确证结果如下:[α]
d24
=+10.8(c 0.98,chcl3)(77%ee)[lit.:[α]
d23
=-14.3(c 1.10,chcl3),(99%ee for(s)-isomer)];ir(film):2926,1680,1453,1219,739cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.07(d,j=8.0hz,1h),7.50-7.43(m,1h),7.35-7.27(m, 3h),7.25-7.18(m,4h),3.50(dd,j=13.5,4.0hz,1h),3.00-2.87(m,2h),2.80-2.71(m, 1h),2.69-2.60(m,1h),2.16-2.07(m,1h),1.86-1.74(m,1h);
13
c nmr(125mhz,cdcl3, ppm)δ199.6,144.2,140.3,133.5,132.7,129.5,128.9,128.6,127.8,126.8,126.3,49.7, 35.9,28.8,27.9;hrms(apci)calcd.for c
17
h
17
o(m+h):237.1274,found:237.1273.
[0191]
经结构鉴定所合成的化合物确为式vi-a所示目标化合物(r)-2-苄基-1-四氢萘酮。
[0192]
实施例22、式vi-b所示化合物的制备
[0193]
按照下述合成路线制备式vi-b所示化合物:
[0194][0195]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-b(e)-2-(4-氯苯亚甲基)-1-四氢萘酮(0.1344 g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为91%, ee值79%。
[0196]
结构确证结果如下:[α]
d21
=+13.7(c 0.87,chcl3)(79%ee);ir(film):2929,1681, 1491,1230,745cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.06(d,j=8.0hz,1h), 7.50-7.42(m,1h),7.36-7.29(m,1h),7.29-7.24(m,2h),7.22(d,j=7.5hz,1h),7.16(d, j=8.0hz,2h),3.49-3.36(m,1h),2.99-2.87(m,2h),2.76-2.62(m,2h),2.14-2.04(m, 1h),1.85-1.72(m,1h);
13
c nmr(125mhz,cdcl3,ppm)δ199.2,144.1,138.7,133.6, 132.6,132.1,130.8,128.9,128.7,127.8,126.9,49.5,35.3,28.9,28.0;hrms(esi)calcd. for c
17
h
14
ocl(m-h):269.0739,found:269.0736.
[0197]
经结构鉴定所合成的化合物确为式vi-b所示目标化合物(r)-2-(4-氯苄基)-1-四氢萘酮。
[0198]
实施例23、式vi-c所示化合物的制备
[0199]
按照下述合成路线制备式vi-c所示化合物:
[0200][0201]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-c(e)-2-(4-甲氧酰基苯亚甲基)-1-四氢萘酮 (0.1462g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为96%,ee值77%。
[0202]
结构确证结果如下:[α]
d25
=+13.6(c 1.00,chcl3)(77%ee);ir(film):2948,1720, 1682,1280,742cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.06(d,j=8.0hz,1h),7.98(d, j=8.0hz,2h),7.53-7.42(m,1h),7.38-7.28(m,3h),7.22(d,j=8.0hz,1h),3.91(s, 3h),3.60-3.46(m,1h),3.04-2.88(m,2h),2.83-2.68(m,2h),2.15-2.03(m,1h),1.87-1.73 (m,1h);
13
c nmr(125mhz,cdcl3,ppm)δ199.1,167.3,145.9,144.1,133.6,132.6, 130.0,129.5,128.9,128.4,127.8,126.9,52.2,49.5,36.0,28.9,28.1;hrms(apci)calcd. for c
19
h
19
o3(m+h):295.1329,found:295.1326.
[0203]
经结构鉴定所合成的化合物确为式vi-c所示目标化合物(r)-2-(4-甲氧酰基苄基)-1
-ꢀ
四氢萘酮。
[0204]
实施例24、式vi-d所示化合物的制备
[0205]
按照下述合成路线制备式vi-d所示化合物:
[0206][0207]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-d(e)-2-(4-甲氧基苯亚甲基)-1-四氢萘酮 (0.1322g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为96%,ee值78%。
[0208]
结构确证结果如下:[α]
d25
=+17.5(c 0.87,chcl3)(78%ee);ir(film):2931,1680, 1512,1246,735cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.06(d,j=7.5hz,1h), 7.50-7.42(m,1h),7.35-7.28(m,1h),7.22(d,j=7.5hz,1h),7.14(d,j=8.5hz,2h), 6.84(d,j=8.5hz,2h),3.79(s,3h),3.40(dd,j=13.5,4.0hz,1h),3.01-2.86(m,2h), 2.75-2.58(m,2h),2.16-2.07(m,1h),1.86-1.70(m,1h);
13
c nmr(125mhz,cdcl3,ppm) δ199.5,158.0,144.0,133.2,132.5,131.9,130.2,128.7,127.5,126.6,113.8,55.2,49.6, 34.7,28.6,27.6;hrms(apci)calcd.for c
18
h
19
o2(m+h):267.1380,found:267.1377.
[0209]
经结构鉴定所合成的化合物确为式vi-d所示目标化合物(r)-2-(4-甲氧苄基)-1-四氢萘酮。
[0210]
实施例25、式vi-e所示化合物的制备
[0211]
按照下述合成路线制备式vi-e所示化合物:
[0212][0213]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-e(e)-2-苯亚甲基-7-甲基-1-四氢萘酮(0.1242 g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为94%, ee值86%。
[0214]
结构确证结果如下:[α]
d27
=+11.8(c 0.80,chcl3)(86%ee);ir(film):2924,1681, 1497,1166,700cm-1
;1h nmr(500mhz,cdcl3,ppm)δ7.88(s,1h),7.34-7.26(m,3h), 7.25-7.18(m,3h),7.11(d,j=8.0hz,1h),3.48(dd,j=14.0,4.0hz,1h),2.96-2.81(m, 2h),2.77-2.69(m,1h),2.67-2.60(m,1h),2.37(s,3h),2.14-2.05(m,1h),1.83-1.69(m, 1h);
13
c nmr(125mhz,cdcl3,ppm)δ199.9,141.4,140.3,136.5,134.5,132.4,129.5, 128.8,128.6,127.8,126.3,49.7,35.9,28.4,28.0,21.2;hrms(apci)calcd.for c
18
h
19
o (m+h):251.1430,found:251.1428.
[0215]
经结构鉴定所合成的化合物确为式vi-e所示目标化合物(r)-2-苄基-7-甲基-1-四氢萘酮。
[0216]
实施例26、式vi-f所示化合物的制备
[0217]
按照下述合成路线制备式vi-f所示化合物,具体操作如下:
[0218][0219]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-f(e)-2-苯亚甲基-7-溴-1-四氢萘酮(0.1566 g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为97%, ee值85%。
[0220]
结构确证结果如下:[α]
d24
=+13.1(c 0.75,chcl3)(85%ee);ir(film):2926,1685, 1403,1210,699cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.18(d,j=2.0hz,1h),7.56 (dd,j=8.0,2.0hz,1h),7.34-7.27(m,2h),7.25-7.19(m,3h),7.10(d,j=8.0hz,1h), 3.47(dd,j=13.5,4.0hz,1h),2.95-2.79(m,2h),2.77-2.69(m,1h),2.68-2.60(m,1h), 2.16-2.06(m,1h),1.84-1.71(m,1h);
13
c nmr(125mhz,cdcl3,ppm)δ198.3,142.9, 139.9,136.2,134.2,130.8,130.6,129.5,128.7,126.5,120.9,49.4,35.8,28.4,27.6;hrms (apci)calcd.for c
17
h
16
obr(m+h):315.0379,found:315.0376.
[0221]
经结构鉴定所合成的化合物确为式vi-f所示目标化合物(r)-2-苄基-7-溴-1-四氢萘酮。
[0222]
实施例27、式vi-g所示化合物的制备
[0223]
按照下述合成路线制备式vi-g所示化合物,具体操作如下:
[0224][0225]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-g(e)-2-苯亚甲基-6-溴-1-四氢萘酮(0.1566 g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为96%, ee值68%。
[0226]
结构确证结果如下:[α]
d27
=+7.2(c 0.78,chcl3)(68%ee);ir(film):2929,1682, 1587,1219,700cm-1
;1h nmr(500mhz,cdcl3,ppm)δ7.92(d,j=8.5hz,1h), 7.49-7.42(m,1h),7.40(s,1h),7.34-7.27(m,2h),7.25-7.19(m,3h),3.48(dd,j=14.0, 4.0hz,1h),2.97-2.84(m,2h),2.79-2.68(m,1h),2.68-2.58(m,1h),2.14-2.04(m,1h), 1.84-1.72(m,1h);
13
c nmr(125mhz,cdcl3,ppm)δ198.7,145.9,140.0,131.8,131.5, 130.3,129.5,129.4,128.7,128.6,126.5,49.5,35.8,28.6,27.6;hrms(apci)calcd.for c
17
h
16
obr(m+h):315.0379,found:315.0376.
[0227]
经结构鉴定所合成的化合物确为式vi-g所示目标化合物(r)-2-苄基-6-溴-1-四氢萘酮。
[0228]
实施例28、式vi-h所示化合物的制备
[0229]
按照下述合成路线制备式vi-h所示化合物:
[0230][0231]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-h(e)-2-苯亚甲基-5-溴-1-四氢萘酮(0.1566 g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为94%, ee值69%。
[0232]
结构确证结果如下:[α]
d26
=+7.4(c 0.82,chcl3)(69%ee);ir(film):2925,1687, 1441,1218,736cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.04(d,j=8.0hz,1h),7.73 (dd,j=8.0,2.0hz,1h),7.34-7.27(m,2h),7.25-7.17(m,4h),3.44(dd,j=14.0,4.5hz, 1h),3.17-3.04(m,1h),2.86-2.70(m,2h),2.69-2.58(m,1h),2.21-2.10(m,1h),1.86-1.72 (m,1h);
13
c nmr(125mhz,cdcl3,ppm)δ198.7,143.1,139.9,137.3,134.7,129.4, 128.7,127.9,127.1,126.5,124.9,48.6,35.6,29.3,26.8;hrms(apci)calcd.for c
17
h
16
obr(m+h):315.0379,found:315.0375.
[0233]
经结构鉴定所合成的化合物确为式vi-h所示目标化合物(r)-2-苄基-5-溴-1-四氢萘酮。
[0234]
实施例29、式vi-i所示化合物的制备
[0235]
按照下述合成路线制备式vi-i所示化合物,具体操作如下:
[0236][0237]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-i(e)-2-(2-甲苯亚甲基)-7-溴-1-四氢萘酮 (0.1636g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为98%,ee值92%。
[0238]
结构确证结果如下:[α]
d26
=-26.7(c 0.72,chcl3)(92%ee);ir(film):2942,1684, 1403,1210,754cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.18(d,j=2.0hz,1h),7.56 (dd,j=8.0,2.0hz,1h),7.19-7.10(m,5h),3.60(dd,j=14.5,4.0hz,1h),2.95-2.79(m, 2h),2.72-2.65(m,1h),2.56-2.50(m,1h),2.33(s,3h),2.14-2.08(m,1h),1.86-1.77(m, 1h);
13
c nmr(125mhz,cdcl3,ppm)δ198.4,142.9,138.2,136.6,136.2,134.2,130.8, 130.7,130.5,130.2,126.6,126.1,120.9,48.3,33.0,28.5,27.8,19.7;hrms(apci)calcd. for c
18
h
18
obr(m+h):329.0536,found:329.0532.
[0239]
经结构鉴定所合成的化合物确为式vi-i所示目标化合物(r)-2-(2-甲基苄基)-7-溴-1
-ꢀ
四氢萘酮。
[0240]
实施例30、式vi-j所示化合物的制备
[0241]
按照下述合成路线制备式vi-j所示化合物:
[0242][0243]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-j(e)-2-(2-乙基苯亚甲基)-7-溴-1-四氢萘酮 (0.1707g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为99%,ee值92%。
[0244]
结构确证结果如下:[α]
d28
=-20.4(c 0.97,chcl3)(92%ee);ir(film):2963,1684, 1472,1210,754cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.19(d,j=2.0hz,1h),7.56 (dd,j=8.5,2.5hz,1h),7.24-7.09(m,5h),3.63(dd,j=14.5,4.0hz,1h),2.94-2.78(m, 2h),2.75-2.60(m,3h),2.57-2.50(m,1h),2.15-2.08(m,1h),1.86-1.76(m,1h),1.23(t,j =7.5hz,3h);
13
c nmr(125mhz,cdcl3,ppm)δ198.4,142.9,142.7,137.4,136.2,134.2, 130.8,130.5,130.3,128.9,126.8,126.0,120.9,49.0,32.3,28.6,27.8,25.7,15.6;hrms (apci)calcd.for c
19
h
20
obr(m+h):343.0692,found:343.0690.
[0245]
经结构鉴定所合成的化合物确为式vi-j所示目标化合物(r)-2-(2-乙基苄基)-7-溴-1
-ꢀ
四氢萘酮。
[0246]
实施例31、式vi-k所示化合物的制备
[0247]
按照下述合成路线制备式vi-k所示化合物:
[0248][0249]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-k(e)-2-(2-甲氧酰基苯亚甲基)-7-溴-1-四氢萘酮(0.1857g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为95%,ee值87%。
[0250]
结构确证结果如下:[α]
d26
=+15.7(c 0.58,chcl3)(87%ee);ir(film):2949,1720, 1684,1280,1110,757cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.17(d,j=2.0hz,1h), 7.98(d,j=8.0hz,2h),7.57(dd,j=8.0,2.0hz,1h),7.29(d,j=8.0hz,2h),7.11(d,j= 8.0hz,1h),3.91(s,3h),3.56-3.46(m,1h),2.95-2.81(m,2h),2.80-2.69(m,2h), 2.13-2.05(m,1h),1.84-1.72(m,1h);
13
c nmr(125mhz,cdcl3,ppm)δ197.8,167.2, 145.5,142.8,136.4,134.1,130.8,130.6,130.0,129.5,128.5,120.9,52.3,49.2,35.9,28.5, 27.8;hrms(apci)calcd.for c
19
h
18
o3br(m+h):373.0434,found:373.0426.
[0251]
经结构鉴定所合成的化合物确为式vi-k所示目标化合物(r)-2-(2-甲氧酰基苄基)-7
-ꢀ
溴-1-四氢萘酮。
[0252]
实施例32、式vi-l所示化合物的制备
[0253]
按照下述合成路线制备式vi-l所示化合物:
[0254][0255]
在手套箱中,室温下,向反应管中加入三(4-氢四氟苯基)硼(0.0229g,0.05mmol),噁唑啉式i-g(0.0245g,0.10mmol),式vii-k(e)-2-(2-甲氧酰基苯亚甲基)-7-溴-1-四氢萘酮(0.1552g,0.5mmol),5ml均三甲苯,在室温下搅拌使其溶解,然后移到高压反应釜中充入氢气(40bar),在30℃下搅拌72小时,旋蒸浓缩,硅胶柱层析提纯,收率为94%,ee值85%。
[0256]
结构确证结果如下:[α]
d24
=+8.6(c 0.79,chcl3)(85%ee);ir(film):2926,1681, 1452,1201,699cm-1
;1h nmr(500mhz,cdcl3,ppm)δ8.32(d,j=1.5hz,1h),7.71 (dd,j=8.0,2.0hz,1h),7.62(d,j=7.5hz,2h),7.53-7.42(m,2h),7.38-7.28(m,4h), 7.27-7.20(m,3h),3.51(dd,j=13.5,4.0hz,1h),3.06-2.87(m,2h),2.83-2.74(m,1h), 2.73-2.64(m,1h),2.19-2.07(m,1h),1.90-1.76(m,1h);
13
c nmr(125mhz,cdcl3,ppm) δ199.6,143.2,140.3,140.2,139.9,133.0,132.0,129.51,129.50,129.1,128.6,127.8,127.2, 126.4,126.1,49.7,35.9,28.5,27.9;hrms(apci)calcd.for c
23
h
21
o(m+h):313.1587, found:313.1584.
[0257]
经结构鉴定所合成的化合物确为式vi-l所示目标化合物(r)-2,7-二苄基-1-四氢萘酮。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1