一种达克替尼中间体的制备方法与流程
1.本发明属于化学制药领域,具体涉及一种达克替尼中间体的制备方法。
背景技术:
2.达克替尼(英文名:dacomitinib)其结构式如下,是美国辉瑞公司(pfizer)研制的第二代、不可逆的egfr酪氨酸激酶抑制剂(tki),该药作用机制类似阿法替尼,能不可逆抑制三种不同erbb家族分子成员,包括egfr(her1),her2和her4。可能因为可以抑制多个erbb家族的蛋白,所以展示出较好的疗效。2018年9月27日获美国fda批准上市,商品名为被批准用于治疗携带egfr基因外显子19缺失或外显子21 l858r置换突变的转移性非小细胞肺癌(nsclc)患者。
[0003][0004]
p05-3a是合成达克替尼的关键中间体。
[0005][0006]
但是,目前合成该中间体的方法报道较少。而达克替尼其他中间体制备方法又存在反应不完全、产生新杂质、合成路线长、总收率较低、分离纯化复杂、成本较高等问题,不利于产业化生产和推广。
技术实现要素:
[0007]
为了解决上述问题,本发明提供了一种达克替尼中间体的制备方法。
[0008]
本发明提供了一种达克替尼中间体的制备方法,它包括如下步骤:
[0009][0010]
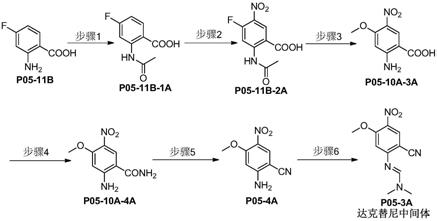
步骤1:将化合物p05-11b、缚酸剂、乙酰化试剂于有机溶剂中反应,得化合物p05-11b-1a;
[0011]
步骤2:在化合物p05-11b-1a中加入硝化剂和脱水剂反应,得化合物p05-11b-2a;
[0012]
步骤3:化合物p05-11b-2a和碱溶于有机溶剂中反应,得到化合物p05-10a-3a;
[0013]
步骤4:化合物p05-10a-3a、制备酰氯的试剂溶于有机溶剂中反应,反应后加入氨水反应,得化合物p05-10a-4a;
[0014]
步骤5:将化合物p05-10a-4a和脱水剂于有机溶剂中反应,得到化合物p05-4a;
[0015]
步骤6:将化合物p05-4a和n,n-二甲基甲酰胺二甲基缩醛于有机溶剂中反应,得达克替尼中间体。
[0016]
进一步地,
[0017]
步骤1中,所述化合物p05-11b、缚酸剂和乙酰化试剂的摩尔比为(1~5):(1~5):(1~5);所述化合物p05-11b和有机溶剂的质量体积比为1:(1~10)(w/v);
[0018]
和/或,步骤2中,所述化合物p05-11b-1a和硝化剂的摩尔比为(1~5):(1~5);所述化合物p05-11b-1a和脱水剂的质量体积比为1:(1~10)(w/v);
[0019]
和/或,步骤3中,所述化合物p05-11b-2a和碱的质量比为(10~20):(1~10);所述化合物p05-11b-2a和有机溶剂的质量体积比为1:(1~20)(w/v);
[0020]
和/或,步骤4中,所述化合物p05-10a-3a和制备酰氯的试剂的质量比为(10~20):(10~20);所述化合物p05-10a-3a和有机溶剂的质量体积比为1:(1~20)(w/v);所述有机溶剂与氨水的体积比为1:(1~100);
[0021]
和/或,步骤5中,所述化合物p05-10a-4a和脱水剂的摩尔比为1:(1~5);所述化合物p05-10a-4a和有机溶剂的质量体积比为1:(1~10)(w/v);
[0022]
和/或,步骤6中,所述化合物p05-4a和n,n-二甲基甲酰胺二甲基缩醛的摩尔比为(1~5):(1~5);所述化合物p05-4a和有机溶剂的质量体积比为1:(1~10)(w/v);
[0023]
优选地,
[0024]
步骤1中,所述化合物p05-11b、缚酸剂和乙酰化试剂的摩尔比为2:4:3;所述化合物p05-11b和有机溶剂的质量体积比为1:5(w/v);
[0025]
和/或,步骤2中,所述化合物p05-11b-1a和硝化剂的摩尔比为1:1;所述化合物
p05-11b-1a和脱水剂的质量体积比为1:6(w/v);
[0026]
和/或,步骤3中,所述化合物p05-11b-2a和碱的质量比为13:5.8;所述化合物p05-11b-2a和有机溶剂的质量体积比为1:10(w/v);
[0027]
和/或,步骤4中,所述化合物p05-10a-3a和制备酰氯的试剂的质量比为15:12.6;所述化合物p05-10a-3a和有机溶剂的质量体积比为1:10(w/v);所述有机溶剂与氨水的体积比为1:1;
[0028]
和/或,步骤5中,所述化合物p05-10a-4a和脱水剂的摩尔比为1:3;所述化合物p05-10a-4a和有机溶剂的质量体积比为1:6(w/v);
[0029]
和/或,步骤6中,所述化合物p05-4a和n,n-二甲基甲酰胺二甲基缩醛的摩尔比为2.3:3.5;所述化合物p05-4a和有机溶剂的质量体积比为1:4(w/v)。
[0030]
进一步地,
[0031]
步骤1中,所述缚酸剂为吡啶、三乙胺、4-二甲氨基吡啶、n,n-二异丙基乙胺、碳酸钾或碳酸钠;所述乙酰化试剂为乙酸酐或乙酰氯;所述有机溶剂为二氯甲烷、四氢呋喃、甲苯、乙酸乙酯或n,n-二甲基甲酰胺;
[0032]
和/或,步骤2中,所述硝化剂为浓硝酸、发烟硝酸或硝酸钾;所述脱水剂为浓硫酸、冰醋酸、乙酸酐或五氧化二磷;
[0033]
和/或,步骤3中,所述碱为氢氧化钠或甲醇钠;所述有机溶剂为甲醇;
[0034]
和/或,步骤4中,所述制备酰氯的试剂为二氯亚砜、草酰氯、三氯化磷或五氯化磷;所述有机溶剂为二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜或乙腈;
[0035]
和/或,步骤5中,所述脱水剂为三氯氧磷、三氟乙酸酐、三聚氯氰、乙酸酐、氯化亚砜、五氧化二磷或三光气;所述有机溶剂为二氯甲烷、四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、乙腈或甲苯;
[0036]
和/或,步骤6中,所述有机溶剂为n,n-二甲基甲酰胺、1,4-二氧六环、四氢呋喃、乙腈或甲苯。
[0037]
进一步地,
[0038]
步骤1中,所述乙酰化试剂的加入方式为滴加;
[0039]
和/或,步骤1中,所述反应为滴加完乙酰化试剂后0~100℃反应1~5h;
[0040]
和/或,步骤2中,所述硝化剂的加入方式为滴加;
[0041]
和/或,步骤2中,所述反应为滴加完硝化剂后0~100℃反应1~5h;
[0042]
和/或,步骤3中,所述反应为20~80℃反应5~10h;
[0043]
和/或,步骤4中,所述化合物p05-10a-3a、制备酰氯的试剂溶于有机溶剂中反应的反应条件为加热回流反应1~5h;
[0044]
和/或,步骤4中,所述加入氨水反应的反应条件为0~60℃反应1~5h;
[0045]
和/或,步骤5中,所述反应为20~150℃搅拌反应1~5h;
[0046]
和/或,步骤6中,所述反应为20-150℃反应1~5h;
[0047]
优选地,
[0048]
步骤1中,所述反应为滴加完乙酰化试剂后室温反应3h;
[0049]
和/或,步骤2中,所述反应为滴加完硝化剂后室温反应2h;
[0050]
和/或,步骤3中,所述反应为40℃反应6h;
[0051]
和/或,步骤4中,所述化合物p05-10a-3a、制备酰氯的试剂溶于有机溶剂中反应的反应条件为加热回流反应2h;
[0052]
和/或,步骤4中,所述加入氨水反应的反应条件为室温反应1h;
[0053]
和/或,步骤5中,所述反应为50℃搅拌反应4h;
[0054]
和/或,步骤6中,所述反应为70℃反应3.5h。
[0055]
进一步地,
[0056]
步骤1中,反应后,将得到的化合物p05-11b-1a提纯,步骤如下:浓缩除去有机溶剂,再倒入冰水中,搅拌,过滤,滤饼用水洗涤,抽干,固体50℃干燥过夜,即得;
[0057]
和/或,步骤2中,反应后,将得到的化合物p05-11b-2a提纯,步骤如下:将反应液倒入冷水中,搅拌,过滤,滤饼用水洗涤,抽干,固体50℃干燥过夜,即得;
[0058]
和/或,步骤3中,反应后,将得到的化合物p05-10a-3a提纯,步骤如下:用浓盐酸调节ph至3,析出固体,过滤,滤饼用水洗涤,抽干,固体50℃干燥过夜,即得;
[0059]
和/或,步骤4中,反应后,将得到的化合物p05-10a-4a提纯,步骤如下:浓缩除去有机溶剂,加水,析出固体,过滤,滤饼依次用2n碳酸氢钠水溶液、水洗涤,抽干,固体50℃干燥过夜,即得;
[0060]
和/或,步骤5中,反应后,将得到的化合物p05-4a提纯,步骤如下:将反应液倒入冰水中,析出固体,过滤,滤饼用水洗涤,抽干,固体50℃干燥过夜,即得;
[0061]
和/或,步骤6中,反应后,将得到的化合物p05-3a提纯,步骤如下:向反应液中加入水,析出固体,过滤,抽干得粗品,粗品中加入乙醇,80℃搅拌1h,降温,过滤,抽干,固体50℃干燥过夜,即得。
[0062]
本发明还提供了一种利用前述的制备方法制备得到的达克替尼中间体制备达克替尼的方法,它包括如下步骤:
[0063][0064]
步骤a中:化合物p05-3a溶于有机溶剂中,通入氢气,在催化量的催化剂作用下反应,得化合物p05-2a;
[0065]
步骤b:将哌啶巴豆酸盐酸盐、碱、缩合剂溶于有机溶剂后,反应,再加入化合物p05-2a,继续反应,得化合物p05-1a;
[0066]
步骤c:将化合物p05-1a、3-氯-4-氟苯胺溶于有机溶剂中,反应,得达克替尼。
[0067]
进一步地,
[0068]
步骤a中,所述通入氢气为常压下通入过量氢气;所述化合物p05-3a和有机溶剂的质量体积比为1:(1~10)(w/v);
[0069]
和/或,步骤b中,所述哌啶巴豆酸盐酸盐、碱、缩合剂和化合物p05-2a的摩尔比为(1~5):(1~5):(1~5):(1~5);所述化合物p05-2a和有机溶剂的质量体积比为1:(1~10)(w/v);
[0070]
和/或,步骤c中,所述化合物p05-1a和3-氯-4-氟苯胺的摩尔比为1:(1~5);所述
化合物p05-1a和有机溶剂的质量体积比为1:(1~10)(w/v);
[0071]
优选地,
[0072]
步骤a中,所述通入氢气为常压下通入过量氢气;所述化合物p05-3a和有机溶剂的质量体积比为1:5(w/v);
[0073]
和/或,步骤b中,所述哌啶巴豆酸盐酸盐、碱、缩合剂和化合物p05-2a的摩尔比为2.5:4.6:3.4:2.3;所述化合物p05-2a和有机溶剂的质量体积比为1:5(w/v);
[0074]
和/或,步骤c中,所述化合物p05-1a和3-氯-4-氟苯胺的摩尔比为1:2;所述化合物p05-1a和有机溶剂的质量体积比为1:5(w/v)。
[0075]
进一步地,
[0076]
步骤a中,所述有机溶剂为甲醇、乙醇、乙酸、乙腈、四氢呋喃或1,4-二氧六环;所述催化剂为镍、铂、钯或铑;优选地,所述催化剂为pto2、pd/c、pd(oh)2或raney ni;更优选地,所述催化剂为5%pd/c;
[0077]
和/或,步骤b中,所述碱为三乙胺、吡啶、n,n-二异丙基乙胺、2,6-二甲基吡啶或n-甲基吗啉;所述缩合剂为1-丙基磷酸环酐、二苯基磷酰氯、叠氮化磷酸二苯酯、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、o-苯并三氮唑-四甲基脲六氟磷酸酯或六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷;所述有机溶剂为二氯甲烷、四氢呋喃、乙腈、n,n-二甲基甲酰胺或二甲亚砜;
[0078]
和/或,步骤c中,所述有机溶剂为冰乙酸、乙腈、甲苯、n,n-二甲基甲酰胺或二甲亚砜。
[0079]
进一步地,
[0080]
步骤a中,所述反应为20~100℃反应5~10h;
[0081]
和/或,步骤a中,反应后,将得到的化合物p05-2a提纯,步骤如下:垫硅藻土过滤,滤液浓缩至干,然后用乙腈打浆,过滤,抽干,固体50℃干燥过夜,即得;
[0082]
和/或,步骤b中,所述将哌啶巴豆酸盐酸盐、碱、缩合剂溶于有机溶剂后反应的反应条件为室温搅拌反应0.5~3h;
[0083]
和/或,步骤b中,所述加入化合物p05-2a后反应的反应条件为0~50℃反应10~15h;
[0084]
和/或,步骤b中,反应后,将得到的化合物p05-1a提纯,步骤如下:将反应液倒入到甲苯和5%氢氧化钠水溶液的混合溶液中,搅拌,过滤,滤饼水和甲苯洗涤,抽干,固体50℃干燥过夜,即得;所述甲苯和5%氢氧化钠水溶液的体积比为1:1;
[0085]
和/或,步骤c中,所述反应为0~50℃反应20~25h;
[0086]
和/或,步骤c中,反应后,将得到的化合物p05提纯,步骤如下:向反应液中加入异丙醇和10%的氢氧化钠水溶液的混合溶液,析出固体,过滤,抽干得粗品,用甲苯打浆,过滤,固体50℃干燥过夜,即得;所述异丙醇和10%的氢氧化钠水溶液的体积比为3:10;
[0087]
优选地,
[0088]
步骤a中,所述反应为30℃反应7h;
[0089]
和/或,步骤b中,所述将哌啶巴豆酸盐酸盐、碱、缩合剂溶于有机溶剂后反应的反应条件为室温搅拌反应0.5h;
[0090]
和/或,步骤b中,所述加入化合物p05-2a后反应的反应条件为室温反应12h;
[0091]
和/或,步骤c中,所述反应为室温反应24h。
[0092]
本发明还提供了一种达克替尼中间体,所述达克替尼中间体的结构式如化合物p05-11b-2a所示:
[0093][0094]
本发明中,v/w代表体积质量比,单位为ml/g;w/v代表质量体积比,单位为g/ml。
[0095]
本发明中,室温为25
±
5℃,过夜为12
±
2h。
[0096]
本发明提供了一种达克替尼关键中间体的新的制备方法,该中间体的合成路线短,反应条件温和、反应时间短,分离纯化方法简单,原料试剂均为常规试剂,成本低,而制备得到的产物收率高,十分利于产业化生产和推广,具有良好的应用前景。
[0097]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0098]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0099]
图1为化合物p05-11b-2a的lcms图谱。
[0100]
图2为化合物p05-3a的hnmr图谱。
[0101]
图3为化合物p05的hnmr图谱。
具体实施方式
[0102]
本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。
[0103]
实施例1、达克替尼中间体(p05-3a)的制备
[0104]
达克替尼中间体(化合物p05-3a)的制备路线如下所示:
[0105][0106]
(1)2-乙酰胺基-4-氟苯甲酸(p05-11b-1a)的合成
[0107]
将2-氨基-4-氟苯甲酸(50g,0.32mol)、三乙胺(64.8g,0.64mol)和二氯甲烷(250ml)加入到反应瓶中,再滴加乙酰氯(37.7g,0.48mol),滴完后室温反应3h,tlc监控反应完全。浓缩除去二氯甲烷,剩余物倒入500ml冰水中,搅拌1h,过滤,滤饼用500ml水洗涤,抽干。收集固体在50℃干燥过夜,得到棕黄色固体(p05-11b-1a)61.4g,96.6%。
[0108]
(2)2-乙酰胺基-4-氟-5-硝基苯甲酸(p05-11b-2a)的合成
[0109]
将2-乙酰胺基-4-氟苯甲酸(20g,0.10mol)溶于浓硫酸(120ml)中,然后在20℃以下滴加浓硝酸(9.84g,0.10mol),滴完后室温反应2h,tlc监控反应完全。将反应液缓慢倒入250ml冷水中,搅拌30min。过滤,滤饼用500ml水洗涤,抽干。收集固体在50℃干燥过夜,得到黄色固体(p05-11b-2a)20.8g,收率84.7%。p05-11b-2a的lcms图谱如图1所示。
[0110]
(3)2-氨基-4-甲氧基-5-硝基苯甲酸(p05-10a-3a)的合成
[0111]
将2-乙酰胺基-4-氟-5-硝基苯甲酸(13g,0.0537mol)、甲醇钠(5.8g,0.107mol)、甲醇(130ml)加入到反应瓶中。加热至40℃反应6h,tlc监控反应完全。降温至室温,用浓盐酸调节ph=3,析出固体。过滤,滤饼用500ml水洗涤,抽干。收集固体在50℃干燥过夜,得到黄色固体(p05-10a-3a)10.5g,收率92.2%。
[0112]
(4)2-氨基-4-甲氧基-5-硝基苯甲酰胺(p05-10a-4a)的合成
[0113]
将2-氨基-4-甲氧基-5-硝基苯甲酸(15g,0.0707mol)、二氯亚砜(12.6g,0.106mol)和二氯甲烷(150ml)加入到反应瓶中,加热回流反应2h,降至室温。将上述反应液滴加到氨水(150ml)中,滴完后室温反应1h,tlc监控反应完全。浓缩除去二氯甲烷,加入150ml水,析出固体。过滤,滤饼依次用2n碳酸氢钠水溶液(150ml)、水(150mlx2)洗涤,抽干。收集固体在50℃干燥过夜,得到黄色固体(p05-10a-4a)13.8g,收率92.7%。
[0114]
(5)2-氨基-4-甲氧基-5-硝基苯甲腈(p05-4a)的合成
[0115]
将2-氨基-4-甲氧基-5-硝基苯甲酰胺(25g,0.12mol)、三氯氧磷(55.2g,0.36mol)和乙腈(150ml)加入到反应瓶中,升温至50℃搅拌反应4h,tlc监控反应完全。将反应液缓慢倒入500ml冰水中,析出固体,过滤,滤饼用200ml水洗涤,抽干。收集固体在50℃干燥过夜,得到黄色固体(p05-4a)20.8g,收率89.7%。
[0116]
(6)n'-(2-氰基-5-甲氧基-4-硝基苯基)-n,n-二甲基甲酰胺(p05-3a)的合成
[0117]
将2-氨基-4-甲氧基-5-硝基苯甲腈(45g,0.23mol)、n,n-二甲基甲酰胺二甲基缩醛(41.7g,0.35mol)和n,n-二甲基甲酰胺(180ml)加到反应瓶中。加热至70℃反应3.5h,tlc监控反应完全。降温至室温,向反应液中加入180ml水,析出固体,过滤,抽干得粗品。向粗品中加入乙醇(500ml),在80℃搅拌1h,降温至室温,过滤,抽干。收集固体在50℃干燥过夜,得到黄色固体(p05-3a)50.6g,收率87.5%。p05-3a的hnmr图谱如图2所示。
[0118]
实施例2、达克替尼的制备
[0119]
利用实施例1制备的达克替尼中间体(p05-3a)制备达克替尼。达克替尼的制备路线如下所示:
[0120][0121]
(a)n'-(2-氰基-5-甲氧基-4-氨基苯基)-n,n-二甲基甲酰胺(p05-2a)的合成
[0122]
将实施例1制备的达克替尼中间体n'-(2-氰基-5-甲氧基-4-硝基苯基)-n,n-二甲基甲酰胺(30g,0.12mol)、5%pd/c(3g)和乙腈(150ml)加入到反应瓶中,常压下通入氢气在30℃反应7h,tlc监控反应完全。垫硅藻土过滤,滤液浓缩至干,然后用60ml乙腈打浆1h,过滤,抽干。收集固体在50℃干燥过夜,得到黄色固体(p05-2a)20g,收率76.3%。
[0123]
(b)(2e)-n-[5-氰基-4-[[(二甲胺基)亚甲基]氨基]-2-甲氧基苯基]-4-(1-哌啶基)-2-丁烯酰胺(p05-1a)的合成
[0124]
在反应瓶中加入乙腈(25ml)、哌啶巴豆酸盐酸盐(5.2g,0.025mol)、n,n-二异丙基乙胺(5.9g,0.046mol)和1-丙基磷酸环酐(11g,0.034mol),室温搅拌反应0.5h,再加入n'-(2-氰基-5-甲氧基-4-氨基苯基)-n,n-二甲基甲酰胺(5g,0.023mol)。然后室温反应12h,tlc监控反应完全。将反应液倒入到甲苯(100ml)和5%氢氧化钠水溶液(100ml)中,搅拌0.5h,过滤,滤饼用50ml水和50ml甲苯洗涤,抽干。收集固体在50℃干燥过夜,得到类白色固体(p05-1a)6.1g,收率72.8%。
[0125]
(c)达克替尼(p05)的合成
[0126]
在反应瓶中加入(2e)-n-[5-氰基-4-[[(二甲胺基)亚甲基]氨基]-2-甲氧基苯基]-4-(1-哌啶基)-2-丁烯酰胺(6g,0.016mol)、3-氯-4-氟苯胺(4.8g,0.032mol)和冰乙酸(30ml),室温反应24h,tlc监控反应完全。向反应液中加入异丙醇(30ml)和10%的氢氧化钠水溶液(100ml),析出固体。过滤,抽干得粗品,用甲苯(60ml)打浆0.5h,过滤,收集固体在50℃干燥过夜,得到类白色固体(达克替尼p05)5.9g,收率77.3%。p05的hnmr图谱如图3所示。
[0127]
综上,本发明提供了一种达克替尼关键中间体的新的制备方法,该中间体的合成路线短,反应条件温和、反应时间短,分离纯化方法简单,原料试剂均为常规试剂,成本低,而制备得到的产物收率高,十分利于产业化生产和推广,具有良好的应用前景。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1