一类2-甲基-4-(1-丙三醇)-呋喃类化合物及其制备方法和应用与流程
本发明涉及放线菌次级代谢产物制备活性化合物领域,具体涉及一类2-甲基-4-(1-丙三醇)-呋喃类化合物及其制备方法和应用。
背景技术:
2-甲基-4-(1-丙三醇)-呋喃(mfpt,结构式如下式(1)),最早于1971年,由日本研究人员从链霉菌mc41-m1和mc340-a1的发酵产物中分离得到。研究表明,该化合物对于疱疹病毒和小鼠淋巴细胞白血病细胞株(l1210)具有抑制作用。因此,该类化合物具有潜在抗癌、抗病毒的功效。
至今,2-甲基-4-(1-丙三醇)-呋喃发现已有40多年,基于该类化合物潜在的药用功效,不断开发结构和功能与mfpt相似的化合物一直是研究人员的主要方向。
放线菌作为一类重要的微生物,以产生种类多样的药理活性物质而广泛受到重视。目前,放线菌仍是先导化合物的主要微生物来源,有多种活性化合物已经在临床得到广泛的应用,如链霉素、红霉素、多氧霉素、金霉素、卡那霉素、氯霉素和庆大霉素等用于临床治疗人的多种疾病。随着对新药开发要求的提高,寻找具有新型作用机制的先导化合物显得十分必要。
但截至目前,还未有报道从放线菌中提取得到天然来源的2-甲基-4-(1-丙三醇)-呋喃类似物。
技术实现要素:
本发明首次从放线菌中提取得到天然来源的2-甲基-4-(1-丙三醇)-呋喃类似物,共三种化合物,大大增加了2-甲基-4-(1-丙三醇)-呋喃类似物的种类。经活性试验发现,该三种2-甲基-4-(1-丙三醇)-呋喃类化合物均可显著抑制hep-g2细胞内脂质堆积,可用于制备降血脂的药物或保健食品。
具体技术方案如下:
一类2-甲基-4-(1-丙三醇)-呋喃类化合物,结构式分别如下式(i)~下式(ⅲ)所示:
式(i)和式(ⅱ)所述的两个化合物为首次将2-甲基-4-(1-丙三醇)-呋喃母核中的5位与另一呋喃环类衍生物加成形成乙醇桥连接形成的二聚体。
式(ⅲ)所述的化合物为首次将2-甲基-4-(1-丙三醇)-呋喃母核与另一吡喃环类衍生物加成形成的二聚体。
显然,上述的三个化合物结构新颖,相较于2-甲基-4-(1-丙三醇)-呋喃的母核结构均已经有了显著变化。
本发明还公开了所述2-甲基-4-(1-丙三醇)-呋喃类化合物的制备方法,包括以下步骤:
1)将放线菌活化后接种于高氏一号液体培养基中,摇床恒温震荡培养,获得发酵液;
所述放线菌,采用中国工业微生物菌种保藏管理中心出售的链霉菌streptomycessp.cicc21933;
2)将所述发酵液经有机溶剂萃取后获得有机萃取液;
3)将所述有机萃取液浓缩后,经分离纯化,分别获得如式(i)~式(ⅲ)结构的2-甲基-4-(1-丙三醇)-呋喃类化合物。
步骤1)中:
所述放线菌活化的过程包括:
将所述放线菌接种到高氏一号琼脂固体培养基上,于28~30℃恒温培养箱中静置,活化培养3~5天。
所述高氏一号液体培养基的配制:以发酵培养基1l为例,可溶性淀粉20g,硝酸钾1g,磷酸氢二钾0.5g,硫酸镁0.5g,硫酸亚铁0.01g,海盐25g加水至1l,调ph7.0~7.2;
所述摇床培养的条件为:28~32℃下,180~200rpm摇床中培养6~10天;更优选的培养条件为28℃下,180rpm摇床中培养8天。
步骤2)中,所述有机溶剂选自乙酸乙酯,经乙酸乙酯萃取后得到的即为乙酸乙酯萃取液。
步骤3)中:
所述浓缩,具体为:
将乙酸乙酯经真空干燥除去溶剂,浓缩处理后的产物继续进行后续的分离纯化步骤。
所述分离纯化,具体为:
(a)浓缩后产物采用反相柱色谱分离,用体积比为1:9至1:0的甲醇-水体系进行梯度洗脱,收集含有目标化合物的馏分并合并;
(b)将步骤(a)所得馏分采用凝胶柱色谱分离,并以纯甲醇作为洗脱液,收集含有目标化合物的馏分并合并;
(c)将步骤(b)所得馏分采用反相高效液相色谱分离纯化,分别得到如式(i)~式(ⅲ)所示的2-甲基-4-(1-丙三醇)-呋喃类化合物。
优选地,步骤(a)中,所述反相柱色谱分离采用的填料为十八烷基键合硅胶。
优选地,步骤(b)中,所述凝胶柱色谱分离采用的填料为羟丙基葡聚糖凝胶。
步骤(c)中,通过分离纯化收集不同洗脱时间的产物即可分别得到式(i)~式(ⅲ)所示的三种化合物。
优选地,采用的检测波长为210nm,采用甲醇体积百分数为10%~100%的甲醇-水体系,以10ml/min进行梯度洗脱40分钟。
收集13.5~14分钟的洗脱液得到如式(i)结构的2-甲基-4-(1-丙三醇)-呋喃类化合物;
收集19.5~20分钟的洗脱液得到如式(ⅱ)结构的2-甲基-4-(1-丙三醇)-呋喃类化合物;
收集7.0~7.5分钟的洗脱液得到如式(ⅲ)结构的2-甲基-4-(1-丙三醇)-呋喃类化合物。
采用上述的制备工艺可以高产率、高纯度的分别获得如式(i)~式(ⅲ)所示的化合物。
为进一步测试本发明分离得到的三种2-甲基-4-(1-丙三醇)-呋喃类化合物的生物活性,采用hepg2细胞进行体外脂质堆积活性评价试验。
试验表明,本发明分离得到的式(i)~式(ⅲ)的三种化合物均能显著的抑制脂质积累,因此可用于制备降血脂的药物或降血脂的保健食品。
与现有技术相比,本发明具有如下优点:
本发明公开了三种2-甲基-4-(1-丙三醇)-呋喃类化合物,均为由放线菌通过发酵培养及提纯后首次得到,其结构为天然产物中首次发现,相较于2-甲基-4-(1-丙三醇)-呋喃的母核结构均已经有了显著变化、结构新颖。
经活性测试发现,三个化合物均能显著的抑制脂质积累,因此可用于制备降血脂的药物或降血脂的保健食品。
附图说明
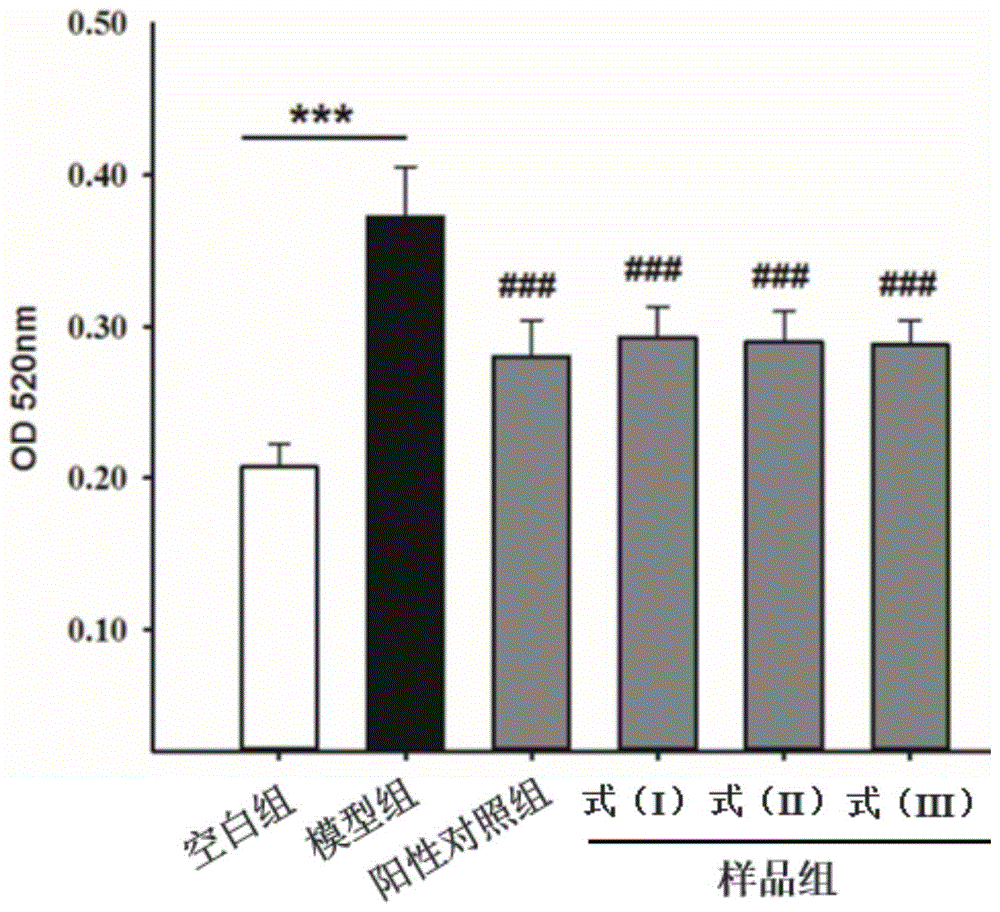
图1为本发明制备的三种2-甲基-4-(1-丙三醇)-呋喃类化合物的降血脂活性的测试数据。
具体实施方式
菌种来源
本发明放线菌采用中国工业微生物菌种保藏管理中心出售的链霉菌streptomycessp.cicc21933。
培养基
高氏一号琼脂固体培养基:以培养基1l计,将可溶性淀粉20g、硝酸钾1g、磷酸氢二钾0.5g、硫酸镁0.5g、硫酸亚铁0.01g、琼脂20g、海盐25g混合后加水至1l,再调节ph值至7.0。
高氏一号液体培养基:以发酵培养基1l计,将可溶性淀粉20g、硝酸钾1g、硫酸氢二钾0.5g、硫酸镁0.5g、硫酸亚铁0.01g、海盐25g混合后加水至1l,再调节ph值至7.0。
实施例1
一、化合物的发酵
1)从原保存的甘油管取放线菌适量,接种到高氏一号琼脂固体培养基上,于30℃培养箱中静置,活化培养3天。
2)将步骤1)中已经活化的放线菌的单菌落接种于500ml锥形瓶中,每瓶含250ml高氏一号液体培养基,在摇床中28℃下以180rpm振荡培养8天,获得发酵液。
二、化合物的制备
(a)将发酵液用等体积的乙酸乙酯萃取3次,所得乙酸乙酯萃取液经旋转蒸发仪减压蒸馏除去乙酸乙酯溶剂得到浓缩液。将所得浓缩液进行反相柱色谱分离,填料为十八烷基键合硅胶,用体积比为1:9至1:0的甲醇-水体系梯度洗脱,薄层检识含有新化合物的馏分,合并。
(b)所得馏分用sephadexlh-20凝胶柱色谱分离,填料为羟丙基葡聚糖凝胶,洗脱剂为甲醇溶液,合并含有新化合物的馏分。
(c)所得含有新化合物的馏分用反相高效液相色谱分离(agilentpursuitc-18(10μm,21.2×250mm)色谱柱,检测波长210nm,采用的流动相为甲醇体积比为10~100%的甲醇-水体系,以10ml/min梯度洗脱40分钟,收集13.5~14min的洗脱液。
将分离纯化得到的化合物进行结构鉴定,分子式根据高分辨质谱hresims计算为c16h22o7([m+na]+349.1260),进一步依据核磁共振数据(表1),其具体结构如下所示,记为furanmycina:
该化合物的核磁共振数据列于下表1中,核磁共振参数1h600mhz,13c150mhz,溶剂dmso-d6。
表1
实施例2
发酵液的获得与实施例1中相同,区别仅在于,化合物的制备步骤(c)中,所得含有新化合物的馏分用反相高效液相色谱分离(agilentpursuitc-18(10μm,21.2×250mm)色谱柱,检测波长210nm,采用的流动相为甲醇体积比为10~100%的甲醇-水体系,以10ml/min梯度洗脱40分钟,收集19.5~20分钟的洗脱液。
将分离纯化得到的化合物进行结构鉴定,分子式根据高分辨质谱hresims计算为c18h24o8([m+na]+391.1365),进一步依据核磁共振数据(表2),其具体结构如下所示,记为furanmycinb:
该化合物的核磁共振数据列于下表2中。核磁共振参数1h600mhz,13c150mhz,溶剂dmso-d6。
表2
实施例3
发酵液的获得与实施例1中相同,区别仅在于,化合物的制备步骤(c)中,所得含有新化合物的馏分用反相高效液相色谱分离(agilentpursuitc-18(10μm,21.2×250mm)色谱柱,检测波长210nm,采用的流动相为甲醇体积比为10~100%的甲醇-水体系,以10ml/min梯度洗脱40分钟,收集7.0~7.5分钟的洗脱液。
将分离纯化得到的化合物进行结构鉴定,分子式根据高分辨质谱hr-esi-ms计算为c16h24o8([m+na]+367.1365),进一步依据核磁共振数据(表3),其具体结构如下所示,记为furanmycinc:
该化合物的核磁共振数据列于下表3中。核磁共振参数1h600mhz,13c150mhz,溶剂dmso-d6。
表3
三、hepg2细胞体外脂质堆积实验
取对数期hepg2细胞加入96孔板中(100μl,约15000个细胞每孔),待70~80%细胞吸附后,将培养基替换成无血清的mem培养基饥饿处理24h。吸弃培养基,实验分为空白组、模型组、阳性对照组、样品组。空白组仅加入无血清的mem培养基,模型组加入含有油酸(100μm)的无血清的mem培养基,阳性对照组加入含有油酸(100μm)及辛伐他丁(10μm)的无血清的mem培养基,样品组加入含有油酸(100μm)及样品式(i)~式(ⅲ)(10μm)的无血清的mem培养基,继续培养24h后,移除培养基,pbs洗一次,用4%多聚甲醛(80μl)固定30分钟后,pbs洗一次,用质量比为60%异丙醇/水处理10分钟后,加入50μl油红工作液(工作液由0.3g/100ml油红/异丙醇的母液与水体积比3:2配制而成)20分钟后,pbs洗三次,异丙醇溶解(70μl)后,在酶标仪520nm波长下测od值。结果见图1(注:与模型组比较,***p<0.001;###p<0.001)。
观察图1可以发现,式(i)~式(ⅲ)的三种化合物可显著抑制hep-g2细胞内脂质堆积,都显示与阳性对照辛伐他丁相当的活性。
可见,本发明为研制新的降血脂药物或保健食品提供了新的天然来源的先导化合物,具有很好的开发应用前景。
- 还没有人留言评论。精彩留言会获得点赞!