本发明属于医药生物技术领域,具体涉及化合物i和ii及其制备方法和在制备抗肿瘤药物中的应用。
背景技术:
癌症正严重威胁着人类的健康。当前,抗肿瘤药物的市场份额也在不断扩大,抗癌药物占医药市场份额高达30%。然而,目前抗癌药物大多存在特异性差、毒副作用强和价格昂贵等缺陷,使得癌症患者常因药物昂贵而放弃治疗或副作用大而意外致死。因此,提高抗癌药物特异性从而降低毒副作用将造福癌症患者,对人类健康和社会发展有着十分重要的现实意义。抗癌药物的主要来源是天然产物及其衍生物。海洋微生物由于代谢产物丰富、生长迅速、需求简单、可再生以及目的化合物产量高等优点,正成为新颖天然药物发现的重要源泉。
技术实现要素:
:
本发明的第一个目的是提供具有抗肿瘤活性的海洋来源杂萜化合物i和ii。
本发明的杂萜化合物,其结构如式(i)中任一所示:
本发明的第二个目的是提供一种化合物i和ii的制备方法,该制备方法中所述的化合物i和ii是从海洋真菌phomopsistersafs441的发酵培养物中分离制备得到的,具体包括以下步骤:
(1)制备海洋真菌phomopsistersafs441的固体发酵培养物,用乙酸乙酯萃取该固体发酵培养物,将乙酸乙酯萃取液经浓缩后得浸膏;
(2)浸膏经过硅胶柱层析,用石油醚-乙酸乙酯以体积比为10:1,5:1,2:1,1:1,0:1和二氯甲烷-甲醇以体积比为10:1,5:1,0:1分别作为洗脱剂,梯度洗脱,收集石油醚-乙酸乙酯体积比5:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=2:1v/v展开得rf=0.3-0.4的组分fr.4;
将组分fr.4经反相柱色谱,用甲醇-水体积比70:30,80:20,90:10,100:0梯度洗脱,收集甲醇-水体积比80:20洗脱组分fr.4.8,收集甲醇-水体积比90:10洗脱组分fr.4.11;
组分fr.4.8经硅胶柱色谱,用二氯甲烷-甲醇以体积比为30:1,20:1,10:1,5:1,2:1作为洗脱剂,梯度洗脱,收集二氯甲烷-甲醇体积比30:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=1:1(v/v)展开得rf=0.4-0.5的组分fr.4.8.1,fr.4.8.1再经硅胶柱色谱,以二氯甲烷-甲醇体积比40:1,30:1,20:1,10:1,5:1作为洗脱剂洗脱,收集二氯甲烷-甲醇体积比30:1洗脱获得的并经tlc薄层层析以正己烷:乙酸乙酯=1:1(v/v)展开得rf=0.4-0.5的组分fr.4.8.1.2,将组分fr.4.8.1.2用hplc分离纯化,得到化合物ii;
组分fr.4.11经凝胶柱色谱,以二氯甲烷-甲醇体积比1:1进行洗脱,洗脱物经tlc薄层层析以正己烷:乙酸乙酯=1:1(v/v)展开得rf=0.3-0.4的组分组分fr.4.11.3用hplc分离纯化,得到化合物i。
所述的制备海洋真菌phomopsistersafs441的固体发酵培养物具体步骤为:挑取fs441的菌丝接种于马铃薯葡萄糖液体培养基中,在28℃、120r/min条件下培养5天,制得种子液,然后将种子液按0.1ml/g的接种量接种于大米培养基中,28℃条件下培养30天,制得fs441的固体发酵培养物,所述的马铃薯葡萄糖液体培养基,每升是通过以下方法配制的:用500ml的纯水煮200g的马铃薯,煮沸20min,过滤得马铃薯汁,再加入葡萄糖20g、kh2po43g、mgso41.5g、维生素b110mg,用水补足至1000ml,灭菌制得;所述的大米培养基是通过以下方法配制的:按每280g大米与300ml质量体积比为0.5%g/ml的粗海盐水溶液混合灭菌制得。
本发明的第三个目的是提供化合物i和/或ii,或其药用盐在制备抗肿瘤药物中的应用。所述的抗肿瘤药物优选为抗肝癌、乳腺癌、神经胶质瘤或非小细胞肺癌药物。
本发明通过实验发现,化合物i和ii对肝癌细胞hepg-2、乳腺癌细胞mcf-7、神经胶质瘤细胞sf-268和非小细胞肺癌细胞a549的ic50值范围为0.5~17.6μm。阳性对照物阿霉素对以上四种肿瘤细胞株的ic50值分别为1.2、1.5、1.1和1.4μm。此结果表明:本发明的化合物i和ii均具有比较显著的抗肿瘤活性。
本发明的第四个目的是提供一种抗肿瘤药物,该抗肿瘤药物包含化合物i和ii中的至少一种,或其药用盐作为活性成分。优选,所述的抗肿瘤药物为抗肝癌、乳腺癌、神经胶质瘤或非小细胞肺癌药物。
本发明的第五个目的是提供海洋真菌phomopsistersafs441在制备化合物i和/或ii中的应用。
与现有技术相比,本发明的优势在于:
本发明从海洋真菌phomopsistersafs441中分离制备得到化合物i和ii,该类化合物i和ii具有比较显著的抗肿瘤活性,可以用于制备抗肿瘤药物,为研究与开发新的抗肿瘤药物提供了候选化合物,为开发利用海洋微生物来源的天然活性物质提供了科学依据。
本发明的海洋真菌phomopsistersafs441已公开于shanchongchen,zhaomingliu,haibotan,yuchanchen,sainili,haohuali,h.guo,shuangzhu,hongxinliu,weiminzhang.tersonea-g,newpyridonealkaloidsfromthedeep-seafungusphomopsistersa.marinedrugs,2019,17,394。该菌株本申请人也持有,并保证自申请日起20年内向公众提供。
附图说明
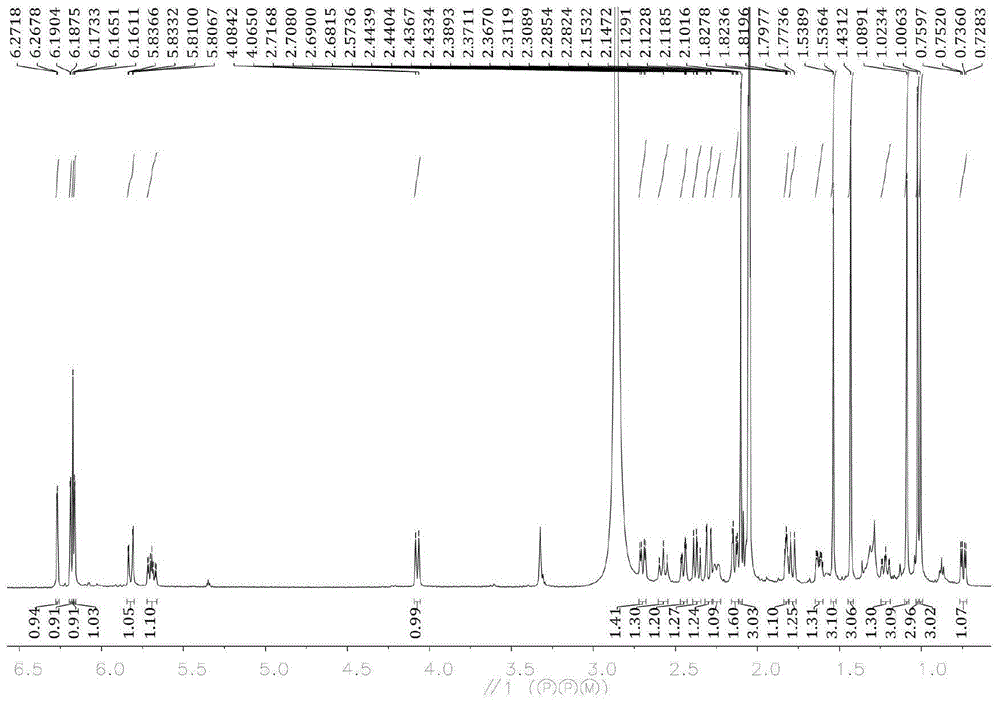
图1是化合物i的1hnmr谱;
图2是化合物i的13cnmr谱;
图3是化合物i的dept谱;
图4是化合物i的hsqc谱;
图5是化合物i的hmbc谱;
图6是化合物i的noesy谱;
图7是化合物i的hr-esims谱;
图8是化合物i的cd谱;
图9是化合物i的uv谱;
图10是化合物i的ir谱;
图11是化合物ii的1hnmr谱;
图12是化合物ii的13cnmr谱;
图13是化合物ii的cosy谱;
图14是化合物ii的hsqc谱;
图15是化合物ii的hmbc谱;
图16是化合物ii的noesy谱
图17是化合物ii的hr-esims谱;
图18是化合物ii的cd谱;
图19是化合物ii的uv谱;
图20是化合物ii的ir谱;
图21是化合物i(1)和ii(2)的1h-1hcosy和hmbc相关图。
具体实施方式
以下实施例是对本发明的进一步说明,而不是对本发明的限制。
实施例1
1、海洋真菌phomopsistersafs441的分离纯化和鉴定
本发明的海洋真菌phomopsistersafs441是2016年5月,从印度洋深海沉积物(88°58.640'n,0°00.307'e,水深3000米)中分离得到的,经its序列分析鉴定,genbank基因登录号为:mk592793,经blast比对,同源分析,鉴定该菌株属于phomopsis属真菌,命名为phomopsistersafs441。
2、phomopsistersafs441的固体发酵
将活化的深海真菌phomopsistersafs441菌丝体接种于马铃薯葡萄糖液体培养基(每升培养基是通过以下方法配制的:用500ml的纯水煮200g的马铃薯,煮沸20min,过滤得马铃薯汁,再加入葡萄糖20g、kh2po43g、mgso41.5g、维生素b110mg,用水补足至1000ml,灭菌制得)中,在28℃、120r/min条件下培养5天,制得种子液,然后将种子液按0.1ml/g大米培养基的接种量接种于大米培养基(该培养基是通过以下方法配制的:将280g大米与300ml质量体积比为0.5%mg/ml的粗海盐水溶液混合,121℃高压灭菌20min,冷却,制得)中,28℃条件下培养30天,制得fs441的固体发酵培养物。
3、化合物i和/或ii的制备
(1)往fs441的固体发酵培养物中加入乙酸乙酯,浸泡提取24小时,重复提取3次,提取液合并后经浓缩后得浸膏(177.9g)。
(2)浸膏经过硅胶柱层析(直径8cm玻璃柱,100-200目硅胶),用石油醚-乙酸乙酯以体积比为10:1,5:1,2:1,1:1,0:1和二氯甲烷-甲醇以体积比为10:1,5:1,0:1分别作为洗脱剂,梯度洗脱,收集石油醚-乙酸乙酯体积比5:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=2:1v/v展开得rf=0.3-0.4的组分fr.4;
将组分fr.4(41.8g)经反相柱色谱(直径3.5cm玻璃柱,填料为rp-c18反相硅胶),用甲醇-水70:30,80:20,90:10,100:0v/v梯度洗脱,得到15个亚组分fr.4.1-fr.4.15。收集甲醇-水80:20v/v洗脱下来的组分fr.4.8,收集甲醇-水体积比90:10洗脱下来的组分fr.4.11;
组分fr.4.8再经硅胶柱色谱(直径2.5cm玻璃柱,100-200目硅胶),用二氯甲烷-甲醇以体积比为30:1,20:1,10:1,5:1,2:1作为洗脱剂,梯度洗脱,收集二氯甲烷-甲醇体积比30:1洗脱获得的且经tlc薄层层析以正己烷:乙酸乙酯=1:1(v/v)展开得rf=0.4-0.5的组分fr.4.8.1,fr.4.8.1再经硅胶柱色谱(直径2.5cm玻璃柱,100-200目硅胶),以二氯甲烷-甲醇体积比40:1,30:1,20:1,10:1,5:1作为洗脱剂洗脱,收集二氯甲烷-甲醇体积比30:1洗脱获得的并经tlc薄层层析以正己烷:乙酸乙酯=1:1(v/v)展开得rf=0.4-0.5的组分fr.4.8.1.2,将组分fr.4.8.1.2经半制备hplc在ymc-ods-a/aq柱,流动相为体积比60:40的乙腈/水,流速为2ml/min,收集保留时间为34.8min的洗脱组分,得到化合物ii。
组分fr.4.11经凝胶柱色谱(sephadexlh-20),以二氯甲烷-甲醇体积比1:1进行洗脱,洗脱的组分经tlc薄层层析以正己烷:乙酸乙酯=1:1(v/v)展开得rf=0.3-0.4的组分fr.4.11.3,fr.4.11.3经半制备hplc,使用ymc-ods-a/aq柱,流动相为体积比60:40的甲醇/水,流速为2ml/min,收集保留时间为40.7min的洗脱组分,得到化合物i。
4、化合物i和ii的结构鉴定
1h-nmr、13c-nmr、hmbc核磁共振谱图用brukeradvance-600核磁共振光谱仪测定,以四甲基硅烷(tms)为内标;esi-ms数据用vgautospec-3000型质谱仪测定;紫外光谱用岛津uv-2600分光光度计测定,其结构鉴定如下:
如图1-21所示,图1是化合物i的1hnmr谱;图2是化合物i的13cnmr谱;图3是化合物i的cosy谱;图4是化合物i的hsqc谱;图5是化合物i的hmbc谱;图6是化合物i的noesy谱;图7是化合物i的hr-esims谱;图8是化合物i的cd谱;图9是化合物i的uv谱;图10是化合物i的ir谱;
图11是化合物ii的1hnmr谱;图12是化合物ii的13cnmr谱;图13是化合物ii的cosy谱;图14是化合物ii的hsqc谱;图15是化合物ii的hmbc谱;图16是化合物ii的noesy谱图17是化合物ii的hr-esims谱;图18是化合物ii的cd谱;图19是化合物ii的uv谱;图20是化合物ii的ir谱。
化合物i为黄色粉末(其核磁数据如表1所示);根据hresims[m–h]–m/z535.2723,c23h33o3,计算值为535.2701,确定化合物的分子式为c32h40o7,不饱和度为13;1h-nmr谱显示一个典型的四取代苯环信号[δh6.16(1h,d,j=2.4hz,h-28),6.27(1h,d,j=2.4hz,h-30)]以及6个甲基氢信号[δh1.43(3h,s,h3-20),1.01(3h,s,h3-21),1.09(3h,s,h3-22),1.02(3h,s,h3-23),1.54(3h,d,j=1.5hz,h3-24),2.10(3h,s,h3-32)]。13c-nmr谱以及dept谱显示有32个碳信号(表2),包括12个季碳(包括6个连氧季碳),9个次甲基,5个亚甲基,以及6个甲基。化合物i的平面结构结构通过进一步分析其cosy、hsqc以及hmbc等二维谱图得以确定(图21),化合物i的绝对构型是通过计算ecd法得以确定。
化合物ii为黄色粉末(其核磁数据如表1所示);根据高分辨质谱(hresims)[m+h]+m/z595.2906,c34h43o9,计算值为595.2902,确定化合物的分子式为c34h42o9,不饱和度为14;通过分析其1d和2d谱图发现化合物ii的谱图与化合物i的谱图具有极大的相似度,化合物ii具有与化合物i相同的b-d环系。进一步通过仔细分析化合物ii的hmbc和cosy相关谱图推测出化合物ii和化合物i之间的差别主要存在于a环和e环上。a环由苯环变成了庚三烯环,e环则为一个furan-2(5h)-one环,通过系统分析其二维谱确定了化合物ii的平面结构。化合物ii的绝对构型是通过分析其noesy谱及计算ecd法进行确定的。
表1化合物i和ii的核磁数据(600mhz/150mhz,δinppm,jinhz)
acd3cocd3.bcdcl3
经过上述方法分离的目标化合物i和ii的结构式如式(i)所示:
实施例2
采用srb法(skehanp,storengr,dominics.newcolorimetriccytotoxicityassayforanticancer-drugscreening[j].jnatlcanceinst,1990,82:1107–1112.)测试化合物i和ii的抗肿瘤活性。
1、试验用试剂:将本发明制备的化合物i和ii用二甲基亚砜(dmso)溶解得浓度为10mg/ml的母液,再用rpmi-1640培养基稀释至所需浓度。阳性对照为顺铂水溶液。
本实验所用肿瘤细胞株为肝癌细胞hepg-2、乳腺癌细胞mcf-7、神经胶质瘤细胞sf-268和非小细胞肺癌细胞a549。
2、实验方法:取对数生长期的hepg-2、mcf-7、sf-268和a549细胞,用胰酶消化,台盼蓝染色计数,台盼蓝排斥实验检测细胞活力大于95%后,用新鲜rpmi-1640培养基调整细胞浓度为3×104个/ml,细胞接种于96孔板,每孔加入180μl的细胞悬液,并设3个空白孔调零,于37℃、5%co2培养箱培养24h。待细胞贴壁后,每孔加入20μl一定浓度的上述化合物化合物i和ii的溶液,阴性对照加20μlrpmi-1640培养基,以阿霉素作阳性对照,每个做三个重复。置于37℃、5%co2培养箱中培养72h后,加入50μl体积分数为50%的冷三氯醋酸水溶液固定细胞,4℃放置1h后用蒸馏水洗涤5次,空气中自然干燥。然后加入由体积分数1%冰醋酸水溶液配制的磺酰罗丹明b(sulforhodamineb,srb)4mg/ml溶液100μl/孔,室温中染色30min,去上清,用1%冰醋酸洗涤5次,空气干燥。最后加入200μl/孔,浓度为10mmol/ml的tris溶液,用酶标仪测定570nm处的吸光值(a),用以下公式计算药物对细胞生长的抑制率:细胞生长抑制率(%)=(1-a样品组/a对照组)×100%。
3、实验结果:本发明制备的化合物i和ii对四株肿瘤细胞的细胞毒性如表2所示。此结果表明:本发明的化合物i和ii具有比较显著的抗肿瘤活性,因此,本发明为研究与开发新的抗肿瘤药物提供了候选化合物,为开发利用深海微生物来源的天然活性物质提供了科学依据。
表2化合物i和ii的对癌细胞的抑制效果
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。