阿糖胞苷4-N位氨基酸衍生物及其制备方法与应用与流程
本发明属于医药技术领域,特别是涉及一类阿糖胞苷4-n位氨基酸衍生物及其制备方法,以及在医药领域的应用。
背景技术:
阿糖胞苷(cytosinearabinoside,ara-c)是胞嘧啶和阿拉伯糖形成的糖苷化合物,是一种细胞周期特异性抗代谢药物,是dna聚合酶的竞争性抑制剂,抑制体内dna生物合成。在临床上,阿糖胞苷主要用于急性白血病的治疗,包括肺癌、消化道癌、头颈部癌及恶性淋巴瘤等。由于阿糖胞苷的4-n位在体内易被肠道黏膜和肝脏部位的胞嘧啶脱氨酶作用脱氨,生成无活性的尿嘧啶阿糖胞苷,导致口服利用度低,因此,需要对其结构进行改进,以期获得更加优良的化合物,特别是对嘧啶环的4-位氨基进行衍生化,可以显著提高活性或生物利用度。例如,首都医科大学的崔国辉等为了提高强极性阿糖胞苷的生物利用度,在4-n上引入一些脂肪酰氨基酰基,提高了其抗宫颈癌细胞hela细胞系的活性;浙江中医药大学的何洁等将阿糖胞苷的4-n与卟啉以共价键连接,合成新的阿糖胞苷卟啉衍生物,获得的衍生物比阿糖胞苷具有更高的光敏抗肿瘤活性。这说明在阿糖胞苷的4位氨基上引入合适的取代基,非常有希望得到更加优良的抗肿瘤衍生物。然而,目前针对阿糖胞苷4-n位氨基酸衍生物的制备方法,特别是开发避免使用硅胶色谱柱分离的合成工艺还鲜有报道。
技术实现要素:
发明目的:针对上述现有技术,本发明提供了一种新的结构改进的阿糖胞苷4-n位氨基酸衍生物,及其制备方法,以及制药应用。
技术方案:本发明所述的阿糖胞苷4-n位氨基酸衍生物,其结构如式i所示:
其中,r表示氨基酸基团。
进一步优选的,r表示天冬氨酸、色氨酸、苏氨酸、缬氨酸、赖氨酸、亮氨酸、异亮氨酸、丙氨酸、苯丙氨酸、精氨酸、甘氨酸、丝氨酸、酪氨酸或脯氨酸。
本发明还公开了上述阿糖胞苷4-n位氨基酸衍生物的制备方法,合成路线如下:
其中,氨基酸3的保护基中,不含有coor1基团,或者保护羧基的r1为苯甲基(bn)或苯甲基衍生物;保护氨基的r2为苄氧羰酰基(cbz)。
具体的,所述制备方法包括以下步骤:
步骤(a),阿糖胞苷1在咪唑催化下用叔丁基二甲基氯硅烷选择性保护糖环上的3位和5位羟基得到化合物2;
步骤(b),化合物2在hobt和edc催化下与保护基保护的氨基酸3发生酰化反应得到氨基酸衍生物4;
步骤(c),化合物4用氢氟酸脱除两个硅醚保护基得到化合物5;
步骤(d),化合物5脱除氨基酸上的保护基,得到全去保护的阿糖胞苷4-n位氨基酸衍生物。
进一步的,步骤(a)中,反应溶剂为dmf;催化剂咪唑用量为阿糖胞苷的2~3倍(摩尔比);叔丁基二甲基氯硅烷试剂为50%wt的甲苯溶液,且叔丁基二甲基氯硅烷的用量为阿糖胞苷的2.05~2.2倍(摩尔比)。反应温度5~15℃,反应时间15~20小时。步骤(a)反应后用15%氯化钠溶液(8~9vol)与乙酸乙酯(12~13vol)进行萃取,且萃取后不需要进行溶剂浓缩,直接进行步骤(b)。
进一步的,步骤(b)中,反应溶剂为dmf;氨基酸用量为阿糖胞苷的1~1.3倍(摩尔比);催化剂hobt用量为阿糖胞苷的1倍(摩尔比);催化剂edc用量为阿糖胞苷的1.2~1.5倍(摩尔比)。反应后用5%碳酸氢钠溶液(10~12vol)与乙酸乙酯(12~13vol)进行萃取,且萃取后不需要进行溶剂浓缩,直接进行步骤(c)。
进一步的,步骤(c)中,反应溶剂为步骤(b)中引入的乙酸乙酯;氢氟酸用量为阿糖胞苷的6~8倍(摩尔比);反应在15~25℃条件下是溶液状态;当降至0~10℃且加入10%碳酸钠溶液(5~7vol)后,反应体系变成悬浊液,产物5沉淀出来。
进一步的,步骤(d)中,将对氨基酸的保护基进行催化氢化脱保护,反应溶剂为乙酸乙酯(20vol)和去离子水(5vol),催化剂10%pd/c的用量为化合物5的0.03~0.05倍(摩尔比),常压氢化。
上述阿糖胞苷4-n位氨基酸衍生物制备抗肿瘤药物的应用也在本发明的保护范围内。
有益效果:本发明通过开发并优化了叔丁基二甲基硅醚对阿糖胞苷的糖环上的3位和5位羟基的选择性保护,然后进行4-n位氨基酸衍生化和硅基脱保护,从而得到了阿糖胞苷4-n位氨基酸衍生物。整条合成工艺路线高效简洁,没有繁琐的硅胶色谱柱分离步骤,具有较强的工业价值。制备所得的一系列阿糖胞苷4-n位氨基酸衍生物可用于抗肿瘤药物的制备。
附图说明
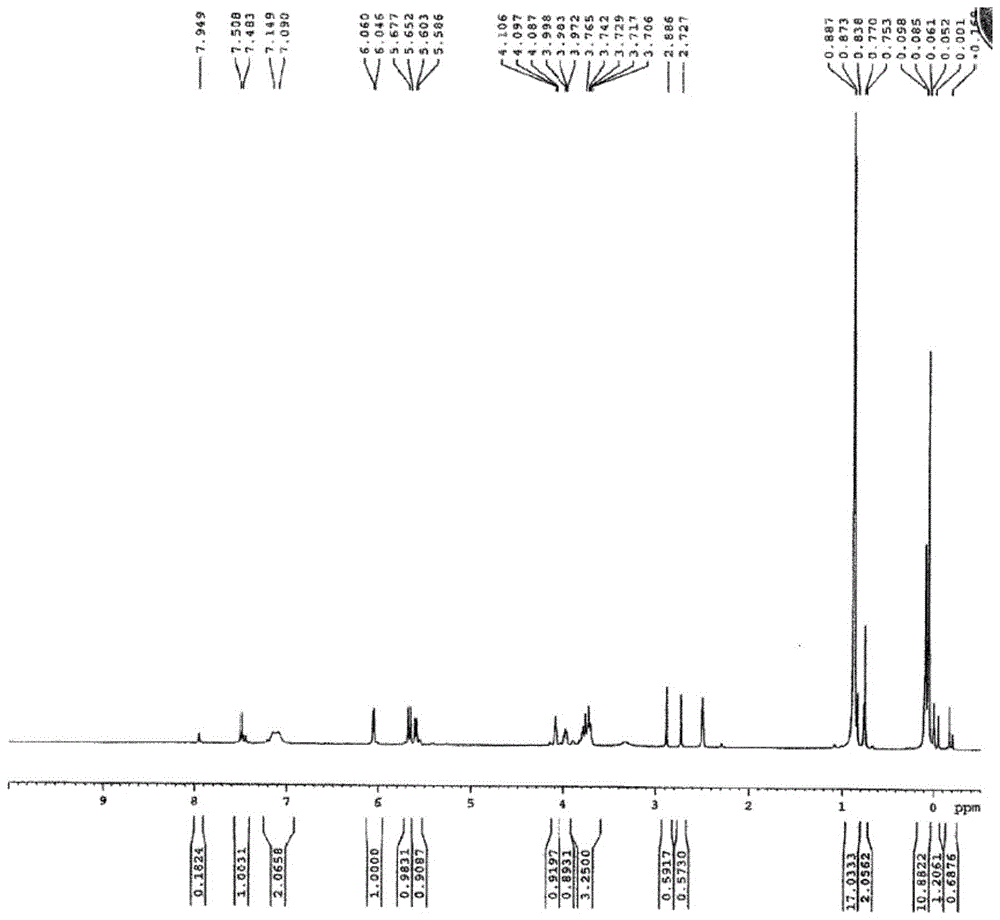
图1是化合物2氢谱;
图2是产物4a氢谱。
图3是产物6a氢谱。
具体实施方式
下面结合具体实施例对本申请作出详细说明。
实施例1
本发明是提供一种阿糖胞苷4-n位天冬氨酸衍生物的制备方法,合成路线如下:
(1)化合物2的合成
将商业化阿糖胞苷(400g,1.65mol,1.0equiv)和咪唑(4.1mol)加入无水dmf(3.6l,9vol)中,反应体系降至-5~5℃,然后加入50%wt叔丁基二甲基氯硅烷(3.54mol)的甲苯溶液,并在5~15℃反应15~20小时。加入15%的氯化钠溶液(8vol)淬灭反应,再加入乙酸乙酯(12vol)进行萃取,收集有机相进行下一步反应。氢谱(400mhz,dmso-d6)见图1。
(2)化合物4a的合成
将化合物3a(1.2equiv),hobthydrate(1.0equiv)和edc(1.3equiv),在室温下加入dmf(vol)体系中。然后通过蠕动泵加入化合物1的乙酸乙酯溶液,滴加时间为30~50分钟。将反应体系在室温下搅拌20~24小时,再加入5%碳酸氢钠溶液(10vol),并用乙酸乙酯(12vol)进行萃取,分离得到乙酸乙酯有机相直接进行下一步反应。氢谱(400mhz,dmso-d6)见图2。其中,反应原料3a的合成路线已报道(carbohydrateresearch,1988,180,147-151)
(3)化合物5a的合成
在化合物4a的乙酸乙酯溶液中于室温下加入氟化氢(7equiv),然后15~25℃条件下搅拌20~24小时。将反应体系温度降至0~10℃,再加入10%碳酸钠溶液(6vol)调制ph值为4.5~5.5。反应体系变成悬浊液,继续搅拌1小时后过滤,用叔丁基甲基醚(10vol)润洗固体,得到化合物5a。
(4)化合物6a的合成
将化合物5a(90g,1.0equiv),乙酸乙酯(20vol)和去离子水(5vol)加入反应瓶中,然后加入4.5g催化剂10wt%pd/c,常压氢化2~3小时。然后过滤反应悬浊液,得到固体产物6a。氢谱(400mhz,dmso-d6)见图3。
实施例2
本发明是提供一种阿糖胞苷4-n位甘氨酸衍生物的制备方法,合成路线如下:
(1)化合物2的合成
将商业化阿糖胞苷(400g,1.65mol,1.0equiv)和咪唑(4.1mol)加入无水dmf(3.6l,9vol)中,反应体系降至-5~5℃,然后加入50%wt叔丁基二甲基氯硅烷(3.54mol)的甲苯溶液,并在5~15℃反应15~20小时。加入15%的氯化钠溶液(8vol)淬灭反应,再加入乙酸乙酯(12vol)进行萃取,收集有机相,进行下一步反应。
(2)化合物4b的合成
将甘氨酸衍生物3b(1.2equiv,),hobthydrate(1.0equiv)和edc(1.3equiv),在室温下加入dmf(vol)体系中。然后通过蠕动泵加入化合物1的乙酸乙酯溶液,滴加时间为30~50分钟。将反应体系在室温下搅拌20~24小时,再加入5%碳酸氢钠溶液(10vol),并用乙酸乙酯(12vol)进行萃取,分离得到乙酸乙酯有机相直接进行下一步反应。
(3)化合物5b的合成
在化合物4b的乙酸乙酯溶液中于室温下加入氟化氢(7equiv),然后15~25℃条件下搅拌20~24小时。将反应体系温度降至0~10℃,再加入10%碳酸钠溶液(6vol)调制ph值为4.5~5.5。反应体系变成悬浊液,继续搅拌1小时后过滤,用叔丁基甲基醚(10vol)润洗固体,得到化合物5b。
(4)化合物6b的合成
将化合物5b(90g,1.0equiv),乙酸乙酯(20vol)和去离子水(5vol)加入反应瓶中,然后加入4.5g催化剂10wt%pd/c,常压氢化2~3小时。然后过滤反应悬浊液,得到固体产物6b。
- 还没有人留言评论。精彩留言会获得点赞!