一种白芨葡甘聚糖双水相提取方法与流程
本发明涉及葡甘聚糖提取技术领域,具体涉及一种白芨葡甘聚糖双水相提取方法。
背景技术:
白芨为兰科植物白芨的茎块,其味苦、甘、涩、性微寒。白芨中富含葡甘聚糖,研究表明白芨葡甘聚糖具有抗炎、促凝血等生物学活性。作为天然高分子材料,白芨葡甘聚糖还有功能缓释性、局部滞留性、自身降解性、无刺激性、无毒副作用廉价易得等特性,在药物制备过程中其应用越来越受到重视。从白芨中提取的中性葡甘聚糖具有明显的清除自由基的能力,随着葡甘聚糖浓度的升高,其清除作用也逐渐增强,呈现量效依赖关系。其清除活性较维生素e高,因此,将白芨胶加入抗衰老营养化妆品中,可补充肌肤营养,消除体内过量的自由基、延缓肌肤衰老。
专利cn201610400993.0一种魔芋葡甘聚糖的提取方法中公开将新鲜的魔芋球茎进行清洗,并去根、芽、皮;再将去皮后的魔芋切成片并放入叠氨钠水溶液中浸泡2~4天;然后将浸泡后的魔芋片研磨成浆液,在浆液中加水并搅拌,直至不再有絮状沉淀产生,然后静置6小时以上,去除底部沉淀,得到魔芋葡甘聚糖水溶液;最后在魔芋葡甘聚糖水溶液中加入乙醇并搅拌,直至不再有沉淀产生,得到魔芋葡甘聚糖乙醇溶液,将魔芋葡甘聚糖乙醇溶液进行减压过滤得到魔芋葡甘聚糖沉淀,使用乙醇冲洗得到高纯度魔芋葡甘聚糖。本专利通过采用叠氨钠,并对水和乙醇进行合理使用,达到提高魔芋葡甘聚糖提取率并保持其本来结构的目的。
目前对于白芨葡甘聚糖的提取方法主要有水提醇沉法、碱水提法、酶解法、超声波进行提取等方法。但是水提法和碱水提法有很多缺点,如耗时长、提取效率低、提取成分不纯等;酶解法增加了工艺的复杂程度,且萃取过程中易造成葡甘聚糖的理化性质、产品纯度和生物活性发生改变。
技术实现要素:
本发明所要解决的技术问题在于提供一种提取效率高、提取质量好的白芨葡甘聚糖双水相提取方法。
本发明通过以下技术手段实现解决上述技术问题的:
一种白芨葡甘聚糖双水相提取方法,包括以下步骤:
(1)将白芨与水混合,于匀浆机中匀浆,制得白芨浆液;
(2)将步骤(1)中的白芨浆液离心,获得上清液;
(3)建立乙醇-磷酸氢二钾双水相体系:乙醇水溶液中乙醇的体积分数为70%,磷酸氢二钾水溶液中磷酸氢二钾的重量分数为55%,所述乙醇水溶液与磷酸氢二钾水溶液的体积比为3:7;
(4)将步骤(2)中的上清液和乙醇-磷酸氢二钾双水相体系置于超声逆流提取装置中提取,所述上清液与乙醇-磷酸氢二钾双水相体系的比为1:45(g:ml);
(5)将步骤(4)中的提取液静置后,取下相溶液过滤,将滤液分别经截留分子量为100k和10k的中空纤维超滤装置进行超滤分离,获得分子量分别为10-100k和大于100k的白芨葡甘聚糖溶液。
有益效果:本发明采用乙醇-磷酸氢二钾双水相提取白芨葡甘聚糖,获得分子量大于100k的白芨葡甘聚糖溶液和分子量为10-100k的白芨葡甘聚糖溶液,制得的白芨葡甘聚糖溶液色泽无色透明,固形物中葡甘聚糖含量95.5-96.3%,蛋白质含量0.32-0.35%,本发明提取周期短、葡甘聚糖提取率高。
优选的,所述步骤(1)中的水为去离子水。
优选的,所述白芨与去离子水的质量比为1:4。
优选的,所述步骤(1)中将白芨与去离子水的混合物置于匀浆机中,于4-10℃、13000-15000rpm、匀浆5min,间歇1min,总匀浆时间为25-30min。
优选的,所述步骤(2)中将白芨浆液于4-10℃、5000-8000rpm、离心15-20min。
有益效果:打碎粗纤维,降低白芨葡甘聚糖粘度,从植物组织中分离出没有溶胀的淀粉颗粒和其他不溶性杂质,利用低温高速匀浆让白芨中的纤维、淀粉和葡甘聚糖解离,再利用低温离心沉淀出没有溶胀的淀粉颗粒和其他不溶性杂质,不添加其他制剂,降低了产品的外源杂质污染;不采用高温处理,均在常温下或低温下进行,降低了温度对白芨葡甘聚糖的破坏和反应。
优选的,所述步骤(4)中于25℃、500w条件下提取20min。
优选的,所述步骤(5)中下相溶液采用400目尼龙滤布过滤。
优选的,所述步骤(5)中于压力为0.20mpa、超滤温度为30℃、料液体积流量为0.06-0.08l/min条件下,截留获得分子量大于100k白芨葡甘聚糖浓缩溶液。
优选的,将经过截留分子量为100k的中空纤维超滤装置超滤后的滤液经过截留分子量为100k的中空纤维超滤装置进行超滤分离,所述步骤(5)中于压力为0.20mpa、超滤温度为25℃、料液体积流量为0.04-0.05l/min条件下,截留获得分子量为10-100k的白芨葡甘聚糖浓缩溶液。
本发明的优点在于:
(1)本发明采用乙醇-磷酸氢二钾双水相提取白芨葡甘聚糖,获得分子量大于100k的白芨葡甘聚糖溶液和分子量为10-100k的白芨葡甘聚糖溶液,制得的白芨葡甘聚糖溶液色泽无色透明,固形物中葡甘聚糖含量95.5-96.3%,蛋白质含量0.32-0.35%;
(2)分子量大于100k、质量分数为2%的白芨葡甘聚糖溶液,粘度范围为362~424cp,ph范围为6.0~6.5,比旋度:左旋28.5~31.0°,干燥失重:≤10%,炽灼残渣:≤1.0%,总灰份:≤1.5%。该白芨葡甘聚糖溶液对络氨酸酶的清除率达到9.72%,对dpph的清除率达到75.22%,在24h内保湿率达到25.6%;
(3)分子量10-100k、质量分数为2%的白芨葡甘聚糖溶液,粘度范围为315~384cp,ph范围为6.0~6.5,比旋度:左旋28.5~31.0°,干燥失重:≤10%,炽灼残渣:≤1.0%,总灰份:≤1.5%。该白芨葡甘聚糖溶液对络氨酸酶的清除率达到9.85%,对dpph的清除率达到78.22%,在24h内保湿率达到22.6%;
(4)本发明提取周期短、葡甘聚糖提取率较高。
附图说明
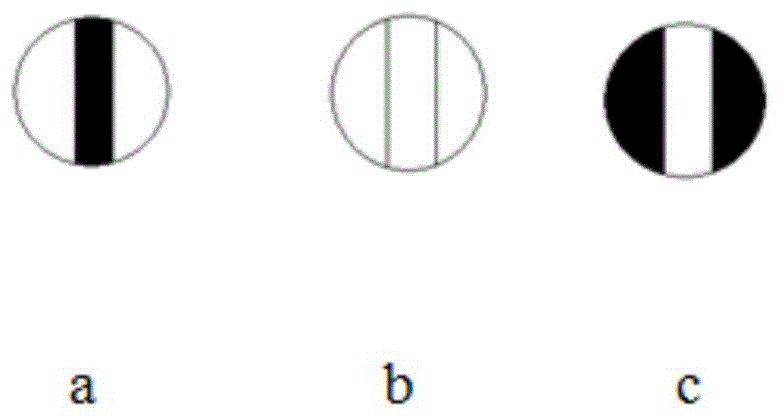
图1为本发明实施例4中旋光仪可观察到的图像。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所用的试验材料和试剂等,如无特殊说明,均可从商业途径获得。
实施例中未注明具体技术或条件者,均可以按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。
实施例1
一种白芨葡甘聚糖双水相提取方法,包括以下步骤:
(1)将白芨洗净后,将白芨与去离子水按质量比为1:4混合,置于匀浆机中,于4℃、13000rpm、匀浆5min,间歇1min,总匀浆时间为30min,制得白芨浆液;
(2)将步骤(1)中的白芨浆液于离心机中,4℃、5000rpm、离心20min,获得上清液;
(3)建立乙醇-磷酸氢二钾双水相体系:乙醇水溶液中乙醇的体积分数为70%,磷酸氢二钾水溶液中磷酸氢二钾的重量分数为55%,乙醇水溶液与磷酸氢二钾水溶液的体积比为3:7;在该体积比条件下,既可以维持稳定的双水相,又可以有效的分离水溶性成分和醇溶性成分;
(4)将步骤(2)中的上清液和乙醇-磷酸氢二钾双水相体系置于超声逆流提取装置中,于25℃、500w条件下提取20min,上清液与乙醇-磷酸氢二钾双水相体系的比为1:45(g:ml);本实施例中采用的超声逆流提取装置为现有技术;
(5)将步骤(4)中的提取液静置后,取下相溶液经400目的尼龙滤布过滤,将滤液经截留分子量为100k的中空纤维超滤装置进行超滤分离,通过超滤装置用去离子水洗去小分子杂质和盐分子,于压力为0.20mpa、超滤温度为30℃、料液体积流量为0.06l/min条件下截留获得分子量大于100k的白芨葡甘聚糖浓缩溶液;检测并控制浓缩溶液的质量分数为20%;
(6)将经过截留分子量为100k的中空纤维超滤装置超滤后的滤液经过截留分子量为100k的中空纤维超滤装置进行超滤分离,通过超滤装置用去离子水洗去小分子杂质和盐分子,于压力为0.20mpa、超滤温度为25℃、料液体积流量为0.04l/min条件下,截留获得分子量为10-100k的白芨葡甘聚糖浓缩溶液,检测并控制浓缩溶液的质量分数为20%。
实施例2
一种白芨葡甘聚糖双水相提取方法,包括以下步骤:
(1)将白芨洗净后,将白芨与去离子水按质量比为1:4混合,置于匀浆机中,于6℃、14000rpm、匀浆5min,间歇1min,总匀浆时间为25min,制得白芨浆液;
(2)将步骤(1)中的白芨浆液于离心机中,6℃、6000rpm、离心15min,获得上清液;
(3)建立乙醇-磷酸氢二钾双水相体系:乙醇水溶液中乙醇的体积分数为70%,磷酸氢二钾水溶液中磷酸氢二钾的重量分数为55%,乙醇水溶液与磷酸氢二钾水溶液的体积比为3:7;在该体积比条件下,既可以维持稳定的双水相,又可以有效的分离水溶性成分和醇溶性成分;
(4)将步骤(2)中的上清液和乙醇-磷酸氢二钾双水相体系置于超声逆流提取装置中,于25℃、500w条件下提取20min,上清液与乙醇-磷酸氢二钾双水相体系的比为1:45(g:ml);本实施例中采用的超声逆流提取装置为现有技术;
(5)将步骤(4)中的提取液静置后,取下相溶液经400目的尼龙滤布过滤,将滤液经截留分子量为100k的中空纤维超滤装置进行超滤分离,于压力为0.20mpa、超滤温度为30℃、料液体积流量为0.07l/min条件下截留获得分子量大于100k的白芨葡甘聚糖浓缩溶液;检测并控制浓缩溶液的质量分数为20%;
(6)将经过截留分子量为100k的中空纤维超滤装置超滤后的滤液经过截留分子量为100k的中空纤维超滤装置进行超滤分离,于压力为0.20mpa、超滤温度为25℃、料液体积流量为0.04l/min条件下,截留获得分子量为10-100k的白芨葡甘聚糖浓缩溶液,检测并控制浓缩溶液的质量分数为20%。
实施例3
一种白芨葡甘聚糖双水相提取方法,包括以下步骤:
(1)将白芨洗净后,将白芨与去离子水按质量比为1:4混合,置于匀浆机中,于10℃、15000rpm、匀浆5min,间歇1min,总匀浆时间为25min,制得白芨浆液;
(2)将步骤(1)中的白芨浆液于离心机中,10℃、8000rpm、离心15min,获得上清液;
(3)建立乙醇-磷酸氢二钾双水相体系:乙醇水溶液中乙醇的体积分数为70%,磷酸氢二钾水溶液中磷酸氢二钾的重量分数为55%,乙醇水溶液与磷酸氢二钾水溶液的体积比为3:7;在该体积比条件下,既可以维持稳定的双水相,又可以有效的分离水溶性成分和醇溶性成分;
(4)将步骤(2)中的上清液和乙醇-磷酸氢二钾双水相体系置于超声逆流提取装置中,于25℃、500w条件下提取20min,上清液与乙醇-磷酸氢二钾双水相体系的比为1:45(g:ml);本实施例中采用的超声逆流提取装置为现有技术;
(5)将步骤(4)中的提取液静置后,取下相溶液经400目的尼龙滤布过滤,将滤液经截留分子量为100k的中空纤维超滤装置进行超滤分离,于压力为0.20mpa、超滤温度为30℃、料液体积流量为0.08l/min条件下截留获得分子量大于100k的白芨葡甘聚糖浓缩溶液,检测并控制浓缩溶液的质量分数为20%;
(6)将经过截留分子量为100k的中空纤维超滤装置超滤后的滤液经过截留分子量为100k的中空纤维超滤装置进行超滤分离,于压力为0.20mpa、超滤温度为25℃、料液体积流量为0.05l/min条件下,截留获得分子量为10-100k的白芨葡甘聚糖浓缩溶液,检测并控制浓缩溶液的质量分数为20%;
实施例4
对实施例1-3中制得的白芨葡甘聚糖溶液进行测定
(一)测定指标:
(1)葡甘聚糖含量:
总糖测定:蒽酮-硫酸比色法;还原糖测定:3,5-二硝基水杨酸比色法;
蛋白含量测定:考马斯亮兰法;葡甘聚糖含量采用总糖减去还原糖的方法来定量;
(2)粘度的测量:采用旋转粘度计测定待测液的粘度;
(3)比旋度的测定:旋光仪的调试:开启旋光仪,预热约5min,当主灯光颜色由淡紫色变为橙黄色,且灯光强度稳定不变时,表示预热完成,可开始调试仪器。
将蒸馏水装入专用玻璃管,盖上玻璃片,盖紧上下盖。防止液体中的气泡影响读数,注意将气泡移至管的突颈处。如图1所示,用蒸馏水进行调试,调节旋光螺旋钮,并观察图像的亮度。将读数盘读数调节到0°处,一边观察亮度一边左右调节旋钮,当三个区域亮度由a变为b时,在继续稍微调节亮度到c,然后往回略调到b(三部分区域亮度相同),表示调节完成。在读数盘上读取此时的旋光度,在测定待测液所得的数据上加上或减去此时的读数。
旋光度的测定:将待测白芨胶液装入专用玻璃管,盖上玻璃片,盖紧上下盖。防止液体中的气泡影响读数,注意将气泡移至管的突颈处。调节旋光螺旋钮,并观察图像的亮度,方法同旋光仪的调试。调节到三个区域亮度相同时,在读数盘上读取此时旋光度,加上或减去调零误差可得到待测白芨胶液的旋光度q。通过计算得到比旋度。
(4)美白性能测试(对络氨酸酶的清除率):向试管中依次加入磷酸盐缓冲溶液、样品溶液、酪氨酸溶液,于35℃水浴10分钟。然后加入酪氨酸酶液,混匀,再在35℃下孵育30min,迅速转移至比色皿中,在475nm处测定吸光值。
受试组用空白对照组1调零,阴性对照组用空白对照组2调零,阳性对照组用空白对照组3调零。
受试组吸光值为a1,阴性对照组吸光值为a2,阳性对照组吸光值为a3。
抑制率=1-[(a1-a2)/(a3-a2)]×100%=(a3-a1)/(a3-a2)×100%。
(5)抗氧化性能测试(对dpph的清除率):在同一个试管加入浓度分别不相同的待检品1ml与120μmol·l-1的dpph配置液3ml,摇晃均匀,避光等待30min,空白组为无水乙醇,在517nm下测吸光度,并按下式计算清除率:
清除率=[a对照-(a样品-a空白)]/a对照×100%
式中:a(对照)为dpph的吸光度(0.534a),a(样品)为加入样品的dpph吸光度,a(空白)为样品本身的吸光度。
(6)保湿能力(称重法)测试:选取医用透气胶带模仿人体肌肤,将医用透气胶带贴在玻璃板上,制成实验装置。在25℃的条件下,准确称量0.5g20%白芨溶液涂布于医用透气胶带的表面,涂抹均匀。随后将其置在有cacl2饱和溶液的干燥器中,经过12h,称量之后的质量,计算其保湿率。
保湿率(100%)=m2/m1×100%
式中:m2为放置在干燥器12h之后样品的质量,g;m1为放置在干燥器前样品的质量g。
(二)测定结果:
(1)分子量大于100k的20%白芨葡甘聚糖溶液色泽无色透明,固形物中葡甘聚糖含量95.5%,蛋白质含量0.35%,熔点为257.2~260℃,其中分子量大于100k的白芨葡甘聚糖溶液的2%溶液粘度范围在362~424cp,ph范围为6.0~6.5;其中固形物的获得方法为于旋转蒸发仪中于60℃蒸发至恒重;
比旋度:左旋28.5~31.0°,干燥失重≤10%,炽灼残渣≤1.0%,总灰份≤1.5%。该白芨提取液对络氨酸酶的清除率达到9.72%,对dpph的清除率达到75.22%,在24小时内保湿率达到25.6%。
分子量10k-100k的20%白芨葡甘聚糖溶液色泽无色透明,固形物中葡甘聚糖含量96.3%,蛋白质含量0.32%,熔点为:257.2~260℃。其中分子量10k-100k的白芨葡甘聚糖溶液的2%溶液粘度范围在:315~384cp,ph范围为:6.0~6.5。比旋度:左旋28.5~31.0°,干燥失重≤10%,炽灼残渣≤1.0%,总灰份≤1.5%。该白芨提取液对络氨酸酶的清除率达到9.85%,对dpph的清除率达到78.22%,在24小时内保湿率达到22.6%。
对比例1
利用去离子水将白芨洗净后,按质量比1:4添加60-90℃的去离子水,置于匀浆机中、13000-15000rpm、匀浆5min,间歇1min、高速匀浆共30-25min;浆液置于离心机中、5000-8000rpm、离心20-15min,分离沉淀,获得上清。
测定方法:对实施例1-3中经过步骤(1)、(2)获得的上清液、本对比例获得的上清液的总粗多糖得率、多糖中淀粉含量进行测定;
其中总粗多糖得率的测定方法采用以下步骤:
根据葡萄糖标准曲线计算出总多浓度,其中葡萄糖标准曲线的测定方法为现有技术,利用下式计算多糖得率:
得率/(mg/g)=c×v1×v2/(m×v3)
式中:c为由标准曲线计算的多糖的浓度,mg/ml;v1为提取液的体积,ml;v2为显色反应的定容体积,ml;v3为测定取样体积,ml;m为样品质量,g。
表1为实施例1-实施例3、对比例中的测定结果
从表1可以看出,对比例1采用热水抽提和常温离心,获得了更高的总粗多糖得率,但是产品中混进了大量的淀粉(葡聚糖),影响了产品(葡甘聚糖)的纯度和品质,而实施例1-实施例3中的低温提取不会造成淀粉溶胀溶解,便于分离。
对比例2
采用专利cn201610400993.0中的方法,将新鲜的白芨块茎清洗去皮后,切成片并放入叠氨钠水溶液中浸泡2~4天;然后将浸泡后的白芨片研磨成浆液,在浆液中加水并搅拌,直至不再有絮状沉淀产生,然后静置6h以上,去除底部沉淀,得到白芨葡甘聚糖水溶液;最后在白芨葡甘聚糖水溶液中加入乙醇并搅拌,直至不再有沉淀产生,得到白芨葡甘聚糖乙醇溶液,将白芨葡甘聚糖乙醇溶液进行减压过滤,得到白芨葡甘聚糖沉淀,使用乙醇冲洗得到白芨葡甘聚糖。
测定结果:
表2为实施例1-实施例3、对比例2中最终产品各指标的测定结果
从表2可以看出,对比例2的提取周期长,产物中混进了大量醇溶性成分的杂质,产品的色泽深,多糖(葡甘聚糖)的萃取率较低,且产品的得率较低,主要杂质蛋白质含量偏高,对络氨酸酶和dpph的清除率低于实施例1-实施例3中制得的葡甘聚糖。
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!