人SGIP1、SCAND3和MYO1G基因甲基化检测试剂盒的制作方法
本发明属于生物
技术领域:
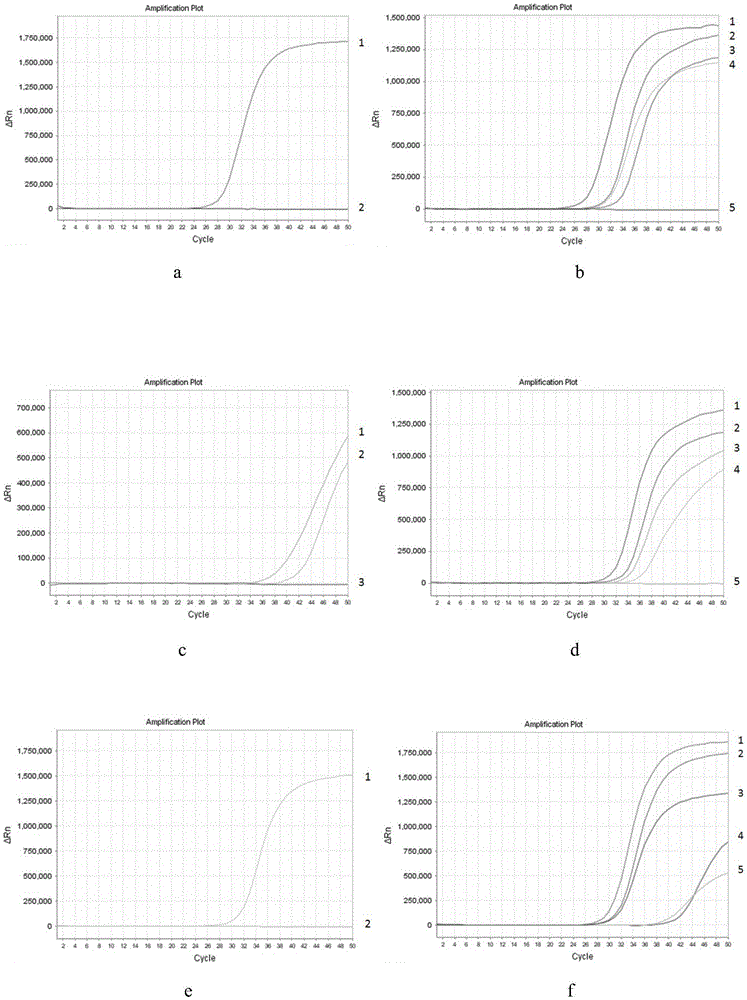
,具体涉及一种人sgip1、scand3和myo1g基因甲基化检测试剂盒。技术背景近年来随着基因检测技术的发展,研究人员发现,除了基因序列改变外,基因的表观遗传学改变也是导致恶性肿瘤发生发展的重要机制(stefanssonoaetal.,theamericanjournalofpathology,2013,183,1052-1063)。dna甲基化是基因表观遗传学改变的主要形式之一。dna甲基化是指在甲基转移酶作用下将甲基转移到胞嘧啶的5位碳原子位置上,形成5-甲基胞嘧啶的过程。大于500bp的dna序列,gc含量大于55%的dna区域被称为cpg岛(suzukimmetal.,naturereviewsgenetics,2008,9,465-476)。哺乳动物50%~60%的启动子区位于cpg岛上(jonespaetal.,naturereviewsgenetics,2009,10,805-811)。正常组织中,cpg岛一般没有甲基化,cpg岛外的cg序列可发生甲基化;当cpg岛的胞嘧啶发生甲基化或cpg岛外的cg序列发生低甲基化时可导致肿瘤的发生(pradhanmpetal.,bmcsystemsbiology,2013,7,141)。研究发现,异常的dna低甲基化可能导致染色体片段丢失或接纳外源染色体,进而导致基因组的不稳定(ballestareetal.,carcinogenesis,2002,23,1103-1109);而在cpg岛的高甲基化状态下,dna呈现扭转状态,染色体螺旋程度增加,双螺旋结构的母链由于侧链的催化产生腔隙,从而导致基因表达的改变(songjetal.,science,2012,335,709-712)。总之,无论是异常的dna高甲基化还是低甲基化均可导致基因表达异常,从而促进肿瘤的发生。鉴于dna异常甲基化与肿瘤发生发展的密切关系,dna甲基化检测对于肿瘤预防、诊断、预后意义重大。scip1是一种能与endophilins相比作用并参与细胞内囊泡运输和受体介导的内吞作用的蛋白(trevaskisjetal.,endocrinology,2005,146,3757-3764)。sgip在物种间广泛保守表达,结构上包含一段n端的与膜相互作用的mp结构域、中部的脯氨酸富裕序列和c端的mu-homolog。人scip1基因包含30个外显子,定位于染色体1p31.3。scip1基因异常甲基化或表达可在恶性肿瘤中被检测到(baejhetal.,oncologyreports,2015,34,1017-1026;songma,etal.,epigenetics,2016,11,464-474)。在包括肺功能、癌症、炎症性疾病和心脏病在内的全基因组研究中,scip1基因甲基化与吸烟有关,吸烟对全基因组甲基化有广泛的影响,且这种影响在戒烟后仍能持续多年(joehanesretal.,circulation:cardiovasculargenetics,2016,9,436-447),提示基因甲基化包括scip1基因甲基化可能是烟草暴露易导致癌症、骨质疏松、肺和心血管疾病等不良健康结果的一种潜在机制。因此,scip1基因甲基化检测或可为肿瘤预防、诊断、预后提供有价值的信息。scand3也被称为zbed9或znf452,是锌指蛋白家族成员之一。scand3结构上包含一个位于n端scan锌指结构域、一个int核心区和一个tpase衍生的hatd二聚化模块(collinstetal.,molecularandcellularbiology,2001,21,3609-3615.)。人scand3基因包含8个外显子,定位于染色体6p22.1。有研究报道,在巴雷特氏食管腺癌中可检测到6p22.1杂合性缺失(wiechtetal.,laboratoryinvestigation,2009,89,385-397)。一项基于中国人群的食管鳞状细胞癌研究发现,scand3基因与食管鳞状细胞癌高风险显著相关(zhangpetal.,plosone,2017,12,e0177494)。在非小细胞肺癌中,scand3可能是akt-gsk3β信号通路的一个上游调节因子,促进肿瘤增殖和侵袭,预示着预后不良(zhangxetal.,oncotarget,2017,8,38863)。此外,cn108753979a公开了一种用于肝癌早期筛查的试剂盒及其使用方法,以scand3为甲基化标志物,用于肝癌诊断,并指出在afp<400μg/l患者中,scand3甲基化发生率高达60%。由此可见,scand3基因甲基化检测具有重要的临床价值。myo1g是一种与质膜相关的i类肌球蛋白(pierceraetal.,thejournalofimmunology,2001,167,3223-3230;patino-lopezgetal.,journalofbiologicalchemistry,2010,285,8675-8686)。人myo1g基因包含22个外显子,定位于染色体7p13。该基因的表达局限于造血细胞(pierceraetal.,thejournalofimmunology,2001,167,3223-3230)。多项研究报道,myo1g基因甲基化与吸烟有关(zeilingersetal.,plosone,2013,8,e63812;besingiwetal.,humanmoleculargenetics,2013,23,2290-2297;elliotthretal.,clinicalepigenetics,2014,6,4;ambatipudisetal.,epigenomics,2016,8,599-618),提示myo1g基因甲基化可能与吸烟诱发的疾病有关。在肺癌中,血液中myo1g基因甲基化可能与肺癌死亡率有关(zhangyetal.,internationaljournalofcancer,2016,139,2482-2492)。在一项针对i期肝细胞癌患者的研究中,研究人员使用包括myo1g基因在内的44个免疫反应相关基因鉴定预后不良高风险患者(nationjb,hepatobiliaryandpancreaticcancerconferenceattheuniversityofhawai‘icancercenter,2018),表明myo1g基因在肝癌预后方面具有一定的临床价值。在乳头状甲状腺癌中,myo1g基因可能作为预测转移和预后的候选生物标志物(zhansetal.,proteomics-clinicalapplications,2019,1900030)。此外,cn108977539a公开了一种用于预测结直肠癌疗效和预后的基因甲基化面板,通过检测结直肠癌血浆中myo1g基因和其它基因甲基化水平的改变以预测结直肠癌的治疗效果、预后和死亡风险。可见,myo1g基因甲基化检测具有重要的临床意义。目前dna甲基化的检测方法主要有基于甲基化敏感限制性内切酶、重亚硫酸盐转化以及亲和富集等原理而建立的一系列方法(lairdpw,naturereviewsgenetics,2010,11,191)。基于甲基化敏感限制性内切酶的dna甲基化检测技术是利用限制性内切酶对所识别序列内甲基化位点敏感性的差异来进行甲基化检测。该技术检测cpg岛甲基化灵敏度高,无需预知被检dna的序列,但是,其缺点在于:①酶切不完全易造成假阳/阴性结果,②限制酶的识别序列有限导致其对dna甲基化位点的检测也有限。基于重亚硫酸盐转化的dna甲基化检测技术是利用重亚硫酸盐对dna进行处理,将未甲基化的胞嘧啶转化为尿嘧啶,然后进行pcr扩增,尿嘧啶转化为胸腺嘧啶,而甲基化的胞嘧啶则不会被重亚硫酸盐修饰,这样dna包含的甲基化信息就转变为具有差异的dna序列,从而实现甲基化检测。该技术对样本纯度、完整性要求较低,不易受拷贝数变化的影响。但是,其缺点在于:①转化过程中样本易降解,导致产物不纯、完整性下降;②转化不完全造成检测结果偏差;③降低了序列复杂度,易遗漏等位基因甲基化,降低了序列杂交的特异性;④操作步骤繁琐,所需试剂种类多,重亚硫酸盐转化后dna还需纯化和回收才能进行后续检测步骤,而dna的纯化和回收会造成dna损失,进一步降低检测的灵敏度。基于亲和富集的dna甲基化检测技术是利用甲基化cpg结合蛋白和5-甲基胞嘧啶抗体富集甲基化片段,从而实现对甲基化的检测。该技术基因组检测速度快,对样本dna纯度、完整性要求低,无需预知被检dna序列。但是,其不足在于:①cpg位点的密度影响富集的效率;②检测甲基化绝对程度时受拷贝数影响较大。目前市场上尚无同步检测人sgip1、scand3和myo1g基因甲基化的试剂盒。技术实现要素:基于此,本发明提供一种人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法),该试剂盒利用甲基化敏感限制性内切酶对于酶切位点甲基化状态的选择性并结合荧光pcr,实现对sgip1、scand3和myo1g三个基因的甲基化状态同步检测,并具有较好的灵敏度。为了实现上述目的,本发明的技术方案如下:人sgip1、scand3和myo1g基因甲基化检测试剂盒,包括甲基化敏感限制性内切酶、酶切缓冲液、荧光pcr反应液,所述甲基化敏感限制性内切酶为aciⅰ、bstuⅰ和hpaⅱ。在其中一些实施例中,所述甲基化敏感限制性内切酶为aciⅰ、bstuⅰ和hpaⅱ混合而成的酶混合液,体积比为1±0.1:1±0.1:1±0.1,每种终浓度均为3.3±0.03u/μl。这种酶混合液的使用可有效避免因酶切不完全造成的假阳/阴性结果。在其中一些实施例中,所述酶切缓冲液中含有bsa,优选地,含有0.1±0.01mg/mlbsa。酶切缓冲液中bsa的添加可增强内切酶的稳定性,并结合dna样本中可能存在的污染物。更优选地,所述酶切缓冲液为含10mmtris-hcl(37℃时ph值7.5)、10mmmgcl2、100mmnacl、0.1mg/mlbsa的水溶液。所述荧光pcr反应液包含pcr缓冲液、dntp、引物及探针、dna聚合酶、bsa、无核酸酶的水;所述引物及探针包含针对sgip1基因的seqidno.1至seqidno.3,针对scand3基因的seqidno.4至seqidno.6,针对myo1g基因的seqidno.7至seqidno.9。在一些实施例中,还包括有酶切内参引物及探针,优选为,序列如seqid10至seqid12所示。在一些实施例中,还包括pcr内参引物及探针,优选为,序列如seqid13至seqid15所示。其中,所有探针的5’端修饰有荧光报告基团,例如fam、vic、rox、cy5或tamara,3’端连接有mgb修饰基团。本发明所述酶切内参引物及探针和pcr内参引物及探针均是以actb基因作为检测靶区而设计的,其中酶切内参引物及探针检测靶区包含aciⅰ、bstuⅰ、hpaⅱ等甲基化敏感限制性内切酶的酶切位点,pcr内参引物及探针检测靶区包含不包含aciⅰ、bstuⅰ、hpaⅱ等甲基化敏感限制性内切酶的酶切位点。这种两组内参引物及探针的设计可有效监控酶切反应情况以及甲基化检测结果的有效性。在其中一些实施例中,所述bsa在荧光pcr反应液中的含量为0.1μg/μl~1μg/μl。bsa的添加能提高pcr反应的效率,同时减少体系中pcr抑制物对pcr反应的影响。在其中一些实施例中,还包括有阴性质控品和阳性质控品。优选地,所述阴性质控品由bsa和非甲基化人类基因组dna组成;所述阳性质控品由bsa、非甲基化人类基因组dna和甲基化人类基因组dna组成。所述阴性质控品和阳性质控品能更好地防止假阳性结果和假阴性结果的产生,从而确保检测结果准确可靠。本发明的另一目的是提供上述试剂盒的使用方法。所述试剂盒的使用方法包括如下步骤:(1)酶切反应:甲基化敏感限制性内切酶和酶切缓冲液配制成酶切反应液处理待测dna样本,酶切反应条件为37℃孵育1小时;酶切反应结束后,加热至85℃,孵育10分钟,对甲基化敏感限制性内切酶进行热灭活,获得酶切产物;(2)荧光pcr反应:向荧光pcr反应液中加入酶切产物进行荧光pcr反应,其荧光pcr反应条件为:95℃预变性5分钟,1个循环;95℃变性30秒,60℃退火30秒,45个循环;40℃降温30s。本发明主要具有以下有益效果:(1)使用甲基化敏感限制性内切酶处理待测dna样本,甲基化核酸序列为甲基化敏感限制性内切酶识别区域,甲基化敏感限制性内切酶对甲基化的核酸序列识别但不酶切,而对非甲基化核酸序列识别并发生酶切活性,从而使甲基化的核酸序列得以完整保留,而非甲基化核酸序列被酶切成小片段;随后使用针对待检区域核酸序列及其包含的甲基化敏感限制性内切酶识别区域设计的位于酶切位点两侧或酶切位点处的特异性引物及位于酶切位点处的探针配制成的荧光pcr反应液对酶切后的待测dna样本进行荧光pcr反应,在此过程中,若待测区域内发生甲基化修饰,则不被酶切能与特异性引物及探针结合,产生荧光信号,若待测区域内未发生甲基化修饰,则被酶切成小片段而不能与特异性引物及探针结合,不产生荧光信号。与基于重亚硫酸盐转化的dna甲基化检测方法相比:需使用重亚硫酸盐,延长产品有效期;避免了因重亚硫酸盐转化造成的dna损失,从而提高检测灵敏度;酶切产物无需纯化和回收步骤可直接进行荧光pcr检测,减少了操作步骤,节省了检测成本和检测时间,避免了因纯化和回收步骤造成的dna损失;特异性引物和探针配合甲基化敏感限制性内切酶酶切位点而设计,引物设计在酶切位点两侧或酶切位点处,探针设计在酶切位点处,待测dna样本被酶切处理后,只有发生甲基化不被酶切的dna才能被pcr扩增并与探针特异性结合产生荧光信号,从而实现了对甲基化序列有效准确的检测。(2)本发明甲基化敏感限制性内切酶为aciⅰ、bstuⅰ和hpaⅱ混合而成的酶混合液,分别识别ccgc、cgcg和ccgg序列,实现对非甲基化核酸中含有的ccgc、cgcg和/或ccgg序列的酶切,可有效提高酶切效率,减少酶切不完全的发生,从而减少假阳性结果的发生,保证检测结果的准确性。(3)本发明可以采用五重荧光pcr反应,能够单管内一次反应检测出sgip1、scand3和myo1g三个基因的甲基化状态,不仅极大地节省了试剂耗材还缩短了检测时间;同时,酶切内参的设计可有效监测酶切反应是否完全,pcr内参的设计可确保甲基化检测结果的有效性,从而确保检测结果准确可靠。此外,本发明试剂盒适用于多种样本类型,可用于血浆dna样本、组织dna样本、细胞dna样本等。(4)进一步地,本发明所述酶切缓冲液含有bsa,不仅可增强内切酶的稳定性以保证酶切效率,还可结合dna样本中可能存在的污染物以防止其对pcr反应的影响。(5)进一步地,本发明所述荧光pcr反应液中添加bsa,不仅能提高pcr反应的效率还能减少体系中pcr抑制物对pcr反应的影响,从而提高检测灵敏度。附图说明图1为实施例2中不同甲基化敏感限制性内切酶用于sgip1、scand3和myo1g基因甲基化检测的荧光pcr扩增曲线图,其中,(a)本发明酶混合液酶切处理非甲基化人类基因组dna(fam、vic、rox和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线,相应ct值在26-27之间(曲线1));(b)本发明酶混合液酶切处理sgip1、scand3和myo1g甲基化人类基因组dna(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线3),vic通道(曲线2),rox通道(曲线4),tamara通道(曲线1));(c)aciⅰ酶切处理非甲基化人类基因组dna(fam、vic和rox通道均无扩增信号,cy5和tamara通道均升起呈s型扩增曲线,cy5通道(曲线2),tamara通道(曲线1));(d)aciⅰ酶切处理sgip1、scand3和myo1g甲基化人类基因组dna(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线3),vic通道(曲线2),rox通道(曲线4),tamara通道(曲线1));(e)bstuⅰ酶切处理非甲基化人类基因组dna(fam、vic、rox和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线(曲线1));(f)bstuⅰ酶切处理sgip1、scand3和myo1g甲基化人类基因组dna(fam、vic、rox、cy5和tamara通道均升起呈s型扩增曲线,fam通道曲线5,vic通道曲线2,rox通道曲线3,cy5通道曲线4,tamara通道ct值为29.64(曲线1));(g)hpaⅱ酶切处理非甲基化人类基因组dna(fam、vic和rox通道均无扩增信号,cy5和tamara通道均升起呈s型扩增曲线,cy5通道曲线2,tamara通道曲线1);(h)hpaⅱ酶切处理sgip1、scand3和myo1g甲基化人类基因组dna(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线1),vic通道(曲线2),rox通道(曲线4),tamara通道(曲线3))。图2为实施例3中本发明与基于重亚硫酸盐转化的甲基化dna检测方法的荧光pcr扩增曲线图。(a)本发明检测非甲基化人类基因组dna(fam、vic、rox和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线,相应ct值在26.5-26.9之间(曲线1));(b)本发明检测sgip1、scand3和myo1g甲基化人类基因组dna(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线1),vic通道(曲线2),rox通道(曲线4),tamara通道(曲线3));(c)基于重亚硫酸盐转化的甲基化dna检测方法检测非甲基化人类基因组dna(没有cy5通道,fam、vic和rox通道均无扩增信号,tamara通道升起呈s型扩增曲线,相应ct值在29-29.6之间(曲线1));(d)基于重亚硫酸盐转化的甲基化dna检测方法检测sgip1、scand3和myo1g甲基化人类基因组dna(没有cy5通道,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线2),vic通道(曲线3),rox通道(曲线4),tamara通道(曲线1))。图3为实施例4中本发明含bsa的荧光pcr反应体系与不添加bsa的荧光pcr反应体系的荧光pcr扩增曲线图。(a)本发明含bsa的荧光pcr反应体系检测非甲基化人类基因组dna(fam、vic、rox和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线,相应ct值在26-26.4之间(曲线1));(b)本发明含bsa的荧光pcr反应体系检测sgip1、scand3和myo1g甲基化人类基因组dna(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线3),vic通道(曲线1),rox通道(曲线4),tamara通道(曲线2));(c)不添加bsa的荧光pcr反应体系检测非甲基化人类基因组dna(fam、vic、rox和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线,相应ct值在29-30.1之间(曲线1));(d)不添加bsa的荧光pcr反应体系检测sgip1、scand3和myo1g甲基化人类基因组dna(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道(曲线2),vic通道(曲线3),rox通道(曲线4),tamara通道(曲线1))。图4为实施例5中本发明检测甲基化dna比例为1%的1ngdna样本的的荧光pcr扩增曲线图(cy5通道无扩增信号,fam、vic、rox和tamara通道均升起呈s型扩增曲线,fam通道曲线2,vic通道曲线3,rox通道曲线4,tamara通道(曲线1))。图5为实施例6中本发明检测个别dna样本的荧光pcr扩增曲线图。(a)健康者dna样本n5的荧光pcr扩增曲线图(fam、vic、rox和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线,相应ct值为27.88(曲线1));(b)sgip1基因甲基化阳性dna样本p1的荧光pcr扩增曲线图(vic、rox和cy5通道均无扩增信号,fam、tamara通道升起呈s型扩增曲线,fam通道ct值为30.74(曲线1),tamara通道ct值为26.81(曲线2));(c)scand3基因甲基化阳性dna样本p4的荧光pcr曲线图;(d)myo1g基因甲基化阳性dna样本p7的荧光pcr曲线图(fam、rox和cy5通道均无扩增信号,vic、tamara通道升起呈s型扩增曲线,vic通道ct值为30.76(曲线2),tamara通道ct值为28.04(曲线1))。图6为本发明阴性质控品的荧光pcr曲线图(rox、fam、vic和cy5通道均无扩增信号,tamara通道升起呈s型扩增曲线,(曲线1))。图7为本发明阳性质控品的荧光pcr曲线图(cy5通道无扩增信号,rox(曲线2)、fam(曲线3)、vic(曲线4)和tamara通道升起呈s型扩增曲线(曲线1))。具体实施方式除非另外指明,本发明的实践将使用分子生物学、细胞生物学、免疫学和重组dna的传统技术,其属于本领域技术范围。参见例如,sambrook,fritsch和maniatis,分子克隆实验指南,第3版(2002)。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例中所用到的各种常用化学试剂,均为市售产品。除非另有定义,本发明所使用的所有的技术和科学术语与属于本发明的
技术领域:
的技术人员通常理解的含义相同。本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不用于限制本发明。本发明所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。下面将结合实施例对本发明进行进一步说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。实施例1:制备本发明的人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法)人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法),包括甲基化敏感限制性内切酶、酶切缓冲液、荧光pcr反应液、阴性参考品和阳性参考品。试剂盒的制备包括以下步骤:(1)甲基化敏感限制性内切酶的制备:根据目的基因选出三种甲基化敏感限制性内切酶,分别为aciⅰ、bstuⅰ和hpaⅱ,按1:1:1的体积比例混合配制成酶混合液,终浓度为每种酶3.3±0.03u/μl,保存备用。所述酶混合液配制方案具体如下:试剂名称每6反应(μl)aciⅰ(10u/μl)1bstuⅰ(10u/μl)1hpaⅱ(10u/μl)1总体积3(2)酶切缓冲液的配制:按照酶切缓冲液的组成进行配制。酶切缓冲液组成如下:10mmtris-hcl(37℃时ph值7.5)、10mmmgcl2、100mmnacl、0.1mg/mlbsa、纯化水。(3)引物与探针的设计及制备:针对sgip1、scand3和myo1g基因的cpg岛分别设计若干特异性引物和荧光探针,将各个引物和探针进行预实验,比较灵敏度、特异性等性能,最终优选出本发明试剂盒的引物及探针,具体如seqid1至seqid9;针对actb基因中包含aciⅰ、bstuⅰ、hpaⅱ等甲基化敏感限制性内切酶的酶切位点的区域设计酶切内参引物及探针,具体如seqid10至seqid12;针对actb基因中不包含aciⅰ、bstuⅰ、hpaⅱ等甲基化敏感限制性内切酶的酶切位点的区域设计pcr内参引物及探针,具体如seqid13至seqid15。所述引物与探针以100μm的母液储存,根据检测需求制备成20μm的工作液备用。所述试剂盒的引物和探针序列具体如下表所示:引物和探针序列(5’-3’)seqidsgip1-f1cgaggccgctcccgtgtgc1sgip1-r1cgctcgagatcgtttctggg2sgip1-p1fam-ggacgcggtggcggcgggactcg-mgb3scand3-f1cctggggaagaggcccgatt4scand3-r1cggaccggaagtgagcgtggc5scand3-p1vic-gcccgtaggttccggccgggcccg-mgb6myo1g-f1ttcgtgcccgctaccgcc7myo1g-r1ggtgacggggctgagcgtg8myo1g-p1rox-acgcggttgtccaatggcggccgg-mgb9actb-f1accgccgagaccgcgtccgcc10actb-r1ccacgatggaggggaagacg11actb-p1cy5-tgatatcgccgcgctcgtcgt-mgb12actb-f2tatgtgggcgacgaggcc13actb-r2gcagcacggggtgctcct14actb-p2tamara-gagcacggcatcgtcaccaact-mgb15(4)荧光pcr反应液的配制:按照本发明的荧光pcr反应液配制方案配制荧光pcr反应液,保存。所述荧光pcr反应液配制方案具体如下:试剂名称每反应(μl)pcr缓冲液5dntp(10mm)2.5引物(20μm)每条加入0.5μl探针(20μm)每条加入0.5μlbsa(10mg/ml)0.25dna聚合酶0.25无核酸酶的水补水至20μl总体积20上述配制体系仅为例证性,在实际应用中可按照比例扩大或缩小该体系体积及其中各组分含量。(5)阴性质控品和阳性质控品的配制:以0.1μgbsa和1ng非甲基化人类基因组dna配制成阴性质控品;以0.1μgbsa、0.9ng非甲基化人类基因组dna和0.1ng甲基化人类基因组dna配制成阳性质控品。(6)试剂盒的分装与组装:试剂盒规格为24人份/盒,分装与组装方案如下:本发明的检测全过程包括以下步骤:(1)按照下表配制酶切反应体系:试剂名称每反应(μl)酶切缓冲液8.5甲基化敏感限制性内切酶0.5待测dna样本1总体积10(2)待测dna样本的酶切反应体系配制好后,混匀离心,置于恒温金属浴中,反应条件设置为37℃反应1小时,85℃灭活10分钟,进行酶切反应,获得酶切产物。(3)以酶切产物为模板,按照下表配制荧光pcr反应体系:试剂名称每反应(μl)荧光pcr反应液20酶切产物5总体积25(4)荧光pcr反应配制好后,混匀离心,放入荧光pcr仪中,设置荧光pcr反应条件为:95℃预变性5分钟,1个循环;95℃变性30秒,60℃退火30秒,45个循环;40℃降温30s。荧光pcr仪荧光通道选择fam、vic、rox、cy5和tamara通道,荧光信号收集是在60℃退火时收集,每个循环收集一次荧光信号。(5)检测结果的判读:根据荧光pcr仪检测到的荧光信号来判读检测结果。检测反应体系的fam、vic、rox、cy5和tamara荧光强度,以cy5来判断酶切反应是否完全,若cy5无扩增信号,说明酶切反应完全,dna样本被完全酶切,若cy5有扩增信号,说明酶切反应不完全;以tamara达到设定的阈值时表示dna上样量在允许范围内,fam、vic、rox信号结果可靠;以fam、vic、rox达到设定阈值时所需要的ct值作为阴阳性判定标准,ct值为0或大于等于45:阴性;ct值小于45:阳性。具体的检测结果判断表如下:实施例2:本发明甲基化敏感限制性内切酶验证实验(1)实验目的本实施例中通过与不同甲基化敏感限制性内切酶的比较,以验证本发明甲基化敏感限制性内切酶的酶切效果。(2)实验方法本实施例中选择非甲基化人类基因组dna、经重亚硫酸盐测序法确定的sgip1、scand3和myo1g甲基化人类基因组dna作为待测样本,分别使用实施例1所述试剂盒中甲基化敏感限制性内切酶(以下表中简称本发明)以及aciⅰ、bstuⅰ、hpaⅱ进行酶切反应,各3个重复,酶切反应体系中加入的dna总量保持一致。酶切反应根据实施例1中的检测步骤进行操作,aciⅰ、bstuⅰ、hpaⅱ按照相应实施例1所述进行酶切操作。酶切产物均使用本发明的荧光pcr反应液根据实施例1中的检测步骤进行荧光pcr检测,酶切产物上样量为5μl。(3)实验结果与分析检测结果如图1和下表所示。从图1和上表检测结果可知,本发明甲基化敏感限制性内切酶处理的dna样本中酶切内参检测结果均呈阴性(ct值>45)(图1a、1b和上表),提示酶切反应是正常和完全的,而aciⅰ、bstuⅰ、hpaⅱ分别处理的dna样本中各有1例酶切内参检测结果呈阳性(ct值<45)(图1c、1f、1g和上表),提示酶切反应不完全,以上说明本发明所述甲基化敏感限制性内切酶的酶切效果较aciⅰ、bstuⅰ、hpaⅱ更佳。本发明所述甲基化敏感限制性内切酶处理的待测dna样本,所有非甲基化人类基因组dna的检测结果皆为甲基化阴性(图1a和上表),所有甲基化人类基因组dna的检测结果皆为甲基化阳性(图1b和上表),说明本发明所述甲基化敏感限制性内切酶检测结果的准确性。与此同时,从图1和上表检测结果可知,本发明所述甲基化敏感限制性内切酶对甲基化人类基因组dna检测的ct值明显低于aciⅰ、bstuⅰ、hpaⅱ,说明本发明甲基化敏感限制性内切酶酶切获得的酶切产物作为模板进行荧光pcr时,其pcr反应体系扩增效率更佳。实施例3:本发明样本前处理方法与重亚硫酸盐转化法比较(1)实验目的本实施例中比较本发明的基于甲基化敏感限制性内切酶消化的甲基化dna检测方法和基于重亚硫酸盐转化的甲基化dna检测方法。(2)实验方法本实施例中选择非甲基化人类基因组dna、经重亚硫酸盐测序法确定的sgip1、scand3和myo1g甲基化人类基因组dna作为待测样本,分别使用本发明的甲基化敏感限制性内切酶和重亚硫酸盐进行待测dna样本前处理,各3个重复。本发明的甲基化敏感限制性内切酶酶切反应根据实施例1中的检测步骤进行操作;重亚硫酸盐转化使用商业化的试剂盒如qiagen公司的epitectfastbisulfiteconversionkits(货号59824)按照其产品说明书进行操作,重亚硫酸盐处理后dna洗脱体积与实施例1所述本发明酶切反应体系体积保持一致,最终获得10μl重亚硫酸盐转化后dna。本发明按照实施例1处理后获得的酶切产物使用本发明的荧光pcr反应液根据实施例1中的检测步骤进行荧光pcr检测,酶切产物上样量为5μl。重亚硫酸盐转化后dna使用根据经重亚硫酸盐处理后的核酸序列设计的特异性引物及探针进行荧光pcr检测,重亚硫酸盐转化后dna上样量为5μl,荧光pcr反应条件与实施例1所述本发明荧光pcr反应条件相同。其中,根据经重亚硫酸盐处理后的核酸序列设计的特异性引物及探针的设计位置与实施例1所述人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法)中的引物及探针设计位置相同;不同在于:①根据经重亚硫酸盐处理后的核酸序列设计的特异性引物及探针序列中的未甲基化胞嘧啶转化成了胸腺嘧啶,②基于重亚硫酸盐转化的甲基化dna检测方法不需要设立酶切内参,因此其引物及探针不包含酶切内参引物及探针。基于重亚硫酸盐转化的甲基化dna检测方法的引物及荧光探针如下:基于重亚硫酸盐转化的甲基化dna检测方法的荧光pcr反应体系如下:试剂名称每反应(μl)pcr缓冲液5dntp(10mm)2.5引物(20μm)每条加入0.5μl探针(20μm)每条加入0.5μlbsa(10mg/ml)0.25dna聚合酶0.25无核酸酶的水补水至20μl重亚硫酸盐转化后dna5总体积25(3)实验结果与分析检测结果如图2和下表所示。从图2和上表检测结果可知,本发明基于甲基化敏感限制性内切酶消化的荧光pcr检测结果与基于重亚硫酸盐转化的荧光pcr检测结果一致,所有非甲基化人类基因组dna样本的检测结果皆为甲基化阴性,所有甲基化人类基因组dna样本的检测结果皆为甲基化阳性;但是,与基于重亚硫酸盐转化的荧光pcr检测的ct值相比,本发明基于甲基化敏感限制性内切酶消化的荧光pcr检测的ct值更低,说明本发明基于甲基化敏感限制性内切酶消化的甲基化检测方法的灵敏度更佳。实施例4:本发明荧光pcr反应液检测效果验证实验(1)实验目的本实施例中通过与不添加bsa的荧光pcr反应体系的检测结果进行对比,验证本发明荧光pcr反应液的检测效果。(2)实验方法本实施例中选择非甲基化人类基因组dna、经重亚硫酸盐测序法确定的sgip1、scand3和myo1g甲基化人类基因组dna作为待测样本,使用实施例1所述试剂盒中的酶切试剂根据实施例中1中的检测步骤进行酶切操作,获得酶切产物。酶切产物分别使用本发明试剂盒中的荧光pcr反应液(含bsa)和不添加bsa的荧光pcr反应体系进行荧光pcr反应,酶切产物上样量均为5μl各3孔。所述试剂盒荧光pcr检测根据实施例1的检测步骤进行,各组荧光pcr反应条件均相同,根据本发明试剂盒的荧光pcr反应条件进行。本发明试剂盒中的荧光pcr反应液含bsa,其配制方案如下:试剂名称每反应(μl)pcr缓冲液5dntp(10mm)2.5引物(20μm)每条加入0.5μl(10条引物共5μl)探针(20μm)每条加入0.5μl(5条探针共2.5μl)bsa(10mg/ml)0.25dna聚合酶0.25无核酸酶的水4.5总体积20本发明试剂盒中的荧光pcr反应液的荧光pcr反应体系:试剂名称每反应(μl)荧光pcr反应液20酶切产物5总体积25不添加bsa的荧光pcr反应体系如下:试剂名称每反应(μl)pcr缓冲液5dntp(10mm)2.5引物(20μm)每条加入0.5μl(10条引物共5μl)探针(20μm)每条加入0.5μl(5条探针共2.5μl)bsa(10mg/ml)0dna聚合酶0.25无核酸酶的水4.75酶切产物5总体积25(3)实验结果与分析检测结果如图3和下表所示。从图3和上表检测结果可知,本发明的荧光pcr反应液与不添加bsa的荧光pcr反应体系的检测结果一致,所有非甲基化人类基因组dna样本的检测结果皆为甲基化阴性,所有甲基化人类基因组dna样本的检测结果皆为甲基化阳性;但是,与不添加bsa的荧光pcr反应体系相比,本发明的荧光pcr反应液检出的ct值更低,说明添加了bsa的本发明荧光pcr反应液检测效果更佳。实施例5:本发明灵敏度分析实验(1)实验目的本实施例中通过对不同甲基化dna比例、不同起始量的dna样本进行检测,以分析本发明的灵敏度。(2)实验方法本实施例中选择非甲基化人类基因组dna、经重亚硫酸盐测序法确定的sgip1、scand3和myo1g甲基化人类基因组dna配制成甲基化dna比例分别为0%、1%、2.5%、5%、10%的起始量分别为10ng、5ng、2ng、1ng、0.1ng的dna样本作为待测样本,使用本实施例1中制备的人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法)根据实施例1中的检测步骤进行酶切和荧光pcr反应。dna样本的配制方案如下:(3)实验结果与分析检测结果如下表所示。从上表检测结果可知,本发明能在dna含量低至1ng,甲基化dna比例低至1%,检测到sgip1、scand3和myo1g基因的甲基化(图4所示),证明了本发明的检测灵敏度高。实施例6:本发明特异性分析实验(1)实验目的本实施例中通过对来自健康者的dna样本、经重亚硫酸盐测序法确定为sgip1、scand3或myo1g甲基化阳性的dna样本进行检测,以分析本发明的特异性。(2)实验方法本实施例中选择来自健康者的dna样本7例、经重亚硫酸盐测序法确定为sgip1、scand3或myo1g甲基化阳性的dna样本各3例作为待测样本,使用本实施例1中制备的人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法)根据实施例1中的检测步骤进行酶切和荧光pcr反应。(3)实验结果与分析检测结果如下表所示(n1-n7代表健康者的dna样本,p1-p3、p4-p6、p7-p9分别代表sgip1、scand3、myo1g甲基化阳性的dna样本);其中,个别dna样本的荧光pcr扩增曲线图如图5所示。从上表检测结果可知,本发明对7份来自健康者的dna样本的检测结果皆为甲基化阴性,3例经重亚硫酸盐测序法确定为sgip1甲基化阳性的dna样本检测结果皆为sgip1甲基化阳性,3例经重亚硫酸盐测序法确定为scand3甲基化阳性的dna样本检测结果皆为scand3甲基化阳性,3例经重亚硫酸盐测序法确定为myo1g甲基化阳性的dna样本检测结果皆为myo1g甲基化阳性,证明了本发明的检测特异性好,阴性符合率为100%,阳性符合率为100%。实施例7:同类产品比较实验(1)实验目的本实施例中通过与甲基化检测“金标准”重亚硫酸盐测序法的检测结果进行对比,验证本发明试剂盒的准确性。(2)实验方法本实施例中收集dna样本共100例作为待测样本,取适量分别使用本发明试剂盒和重亚硫酸盐测序法进行检测。本发明试剂盒检测过程根据实施例1中的检测步骤进行。重亚硫酸盐测序法委托美吉生物完成。(3)实验结果与分析检测结果如下表所示。从上表检测结果可知,本发明试剂盒的检测结果与重亚硫酸盐测序法检测结果的符合率为100%,证明了本发明的检测准确性。实施例8:不同dna样本类型检测结果对比实验(1)实验目的本实施例中通过对不同类型dna样本的检测,以分析本发明试剂盒适用的样本类型。(2)实验方法本实施例中收集20例肿瘤患者的血浆dna样本、组织dna样本、细胞dna样本各1份作为待测样本,使用本实施例1中制备的人sgip1、scand3和myo1g基因甲基化检测试剂盒(荧光pcr法)根据实施例1中的检测步骤进行酶切和荧光pcr反应。(3)实验结果与分析检测结果如下表所示。从上表检测结果可知,对于同时提供血浆dna、组织dna和细胞dna的20例肿瘤患者,三种dna样本检测结果完全相同,符合率为100%,说明本发明试剂盒适用于多种dna样本类型,包括血浆dna、组织dna和细胞dna。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。序列表<110>益善生物技术股份有限公司<120>人sgip1、scand3和myo1g基因甲基化检测试剂盒<160>27<170>siposequencelisting1.0<210>1<211>19<212>dna<213>人工序列(artificialsequence)<400>1cgaggccgctcccgtgtgc19<210>2<211>20<212>dna<213>人工序列(artificialsequence)<400>2cgctcgagatcgtttctggg20<210>3<211>23<212>dna<213>人工序列(artificialsequence)<400>3ggacgcggtggcggcgggactcg23<210>4<211>20<212>dna<213>人工序列(artificialsequence)<400>4cctggggaagaggcccgatt20<210>5<211>21<212>dna<213>人工序列(artificialsequence)<400>5cggaccggaagtgagcgtggc21<210>6<211>24<212>dna<213>人工序列(artificialsequence)<400>6gcccgtaggttccggccgggcccg24<210>7<211>18<212>dna<213>人工序列(artificialsequence)<400>7ttcgtgcccgctaccgcc18<210>8<211>19<212>dna<213>人工序列(artificialsequence)<400>8ggtgacggggctgagcgtg19<210>9<211>24<212>dna<213>人工序列(artificialsequence)<400>9acgcggttgtccaatggcggccgg24<210>10<211>21<212>dna<213>人工序列(artificialsequence)<400>10accgccgagaccgcgtccgcc21<210>11<211>20<212>dna<213>人工序列(artificialsequence)<400>11ccacgatggaggggaagacg20<210>12<211>21<212>dna<213>人工序列(artificialsequence)<400>12tgatatcgccgcgctcgtcgt21<210>13<211>18<212>dna<213>人工序列(artificialsequence)<400>13tatgtgggcgacgaggcc18<210>14<211>18<212>dna<213>人工序列(artificialsequence)<400>14gcagcacggggtgctcct18<210>15<211>22<212>dna<213>人工序列(artificialsequence)<400>15gagcacggcatcgtcaccaact22<210>16<211>19<212>dna<213>人工序列(artificialsequence)<400>16cgaggtcgttttcgtgtgc19<210>17<211>20<212>dna<213>人工序列(artificialsequence)<400>17cgctcgaaatcgtttctaaa20<210>18<211>23<212>dna<213>人工序列(artificialsequence)<400>18ggacgcggtggcggcgggattcg23<210>19<211>20<212>dna<213>人工序列(artificialsequence)<400>19tttggggaagaggttcgatt20<210>20<211>21<212>dna<213>人工序列(artificialsequence)<400>20cgaaccgaaaataaacgtaac21<210>21<211>24<212>dna<213>人工序列(artificialsequence)<400>21gttcgtaggtttcggtcgggttcg24<210>22<211>18<212>dna<213>人工序列(artificialsequence)<400>22ttcgtgttcgttatcgtt18<210>23<211>19<212>dna<213>人工序列(artificialsequence)<400>23aataacgaaactaaacgta19<210>24<211>24<212>dna<213>人工序列(artificialsequence)<400>24acgcggttgtttaatggcggtcgg24<210>25<211>18<212>dna<213>人工序列(artificialsequence)<400>25tatgtgggcgacgaggtt18<210>26<211>18<212>dna<213>人工序列(artificialsequence)<400>26acaacacgaaatactcct18<210>27<211>22<212>dna<213>人工序列(artificialsequence)<400>27gagtacggtatcgttattaatt22当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1